Testes pré-natais e genética reprodutiva
Os avanços na genética reprodutiva e nos testes pré-natais estão revolucionando a maneira como diagnosticamos e tratamos condições genéticas fetais, oferecendo novas esperanças para pais e profissionais de saúde. Com técnicas como o teste pré-natal não invasivo (NIPT) e o diagnóstico pré-natal não invasivo (NIPD), podemos identificar trissomias cromossômicas e doenças genéticas específicas de forma rápida e precisa, permitindo intervenções médicas precoces e aconselhamento genético individualizado. Além disso, o sequenciamento rápido pré-natal do exoma está oferecendo uma análise abrangente do genoma fetal em um curto espaço de tempo, possibilitando diagnósticos precisos e tratamentos direcionados para condições genéticas complexas durante a gestação.
Existem diversas técnicas disponíveis para diagnosticar anomalias fetais e distúrbios genéticos. Entre elas estão exames não invasivos, como o teste combinado no soro materno e ultrassonografias, que podem detectar anormalidades estruturais. Para diagnósticos mais precisos, são utilizadas técnicas invasivas, como amniocentese e amostragem de vilosidades coriônicas. A colaboração com geneticistas clínicos tornou-se crucial nesse processo, uma vez que avanços significativos na medicina têm permitido diagnósticos pré-natais mais detalhados, possibilitando a detecção precoce de anomalias genéticas e fetais.
Técnicas utilizadas no diagnóstico pré-natal
Tabela 1. Técnicas padrão utilizadas no diagnóstico pré-natal
| Técnica | Tempo ideal (semanas) | Transtornos Diagnosticados |
| Não invasivo | ||
| EXAME DE SORO MATERNO | ||
| Teste combinado | 10–14 | Síndrome de Down, síndrome de Edwards, síndrome de Patau |
| Ultrassom | 18–20 | Anormalidades estruturais (por exemplo, sistema nervoso central, coração, rins, membros) |
| Invasivo | ||
| Amniocentese | 15+ | Anormalidades cromossômicas, distúrbios metabólicos, defeitos moleculares |
| Amostragem de vilosidades coriônicas | 11–13 + 6 | Anormalidades cromossômicas, distúrbios metabólicos, defeitos moleculares |
| Fetoscopia (raramente usada no diagnóstico pré-natal) -> Síndrome de Transfusão Feto-Fetal (STFF) | ||
| Sangue (cordocentese) | 18 - 20 | Anomalias cromossômicas, distúrbios hematológicos, infecção congênita |
| Fígado – Biópsia hepática | Distúrbios metabólicos (por exemplo, deficiência de ornitina transcarbamilase) | |
| Pele - Biópsia | Distúrbios hereditários da pele (por exemplo, epidermólise bolhosa) | |
Ultrassonografia
A ultrassonografia (USS) é útil não apenas para indicações obstétricas, como localização placentária e diagnóstico de gestações múltiplas, mas também para avaliação do tamanho fetal e diagnóstico pré-natal de anormalidades estruturais. Não é invasivo e não apresenta risco conhecido para o feto ou a mãe. Equipamentos altamente avançados nas mãos de um operador qualificado e experiente são cada vez mais sensíveis. Por exemplo, pode ser detectada polidactilia, que pode fazer parte de uma síndrome de anormalidades múltiplas, como uma das síndromes autossômicas recessivas de polidactilia de costelas curtas associada à hipoplasia pulmonar grave – muitas vezes letal (Fig. 1 ). Da mesma forma, uma varredura pode revelar que o feto tem uma mandíbula pequena, o que pode estar associado a uma fenda palatina posterior e outras anormalidades mais graves em diversas síndromes específicas (Fig. 2 ).
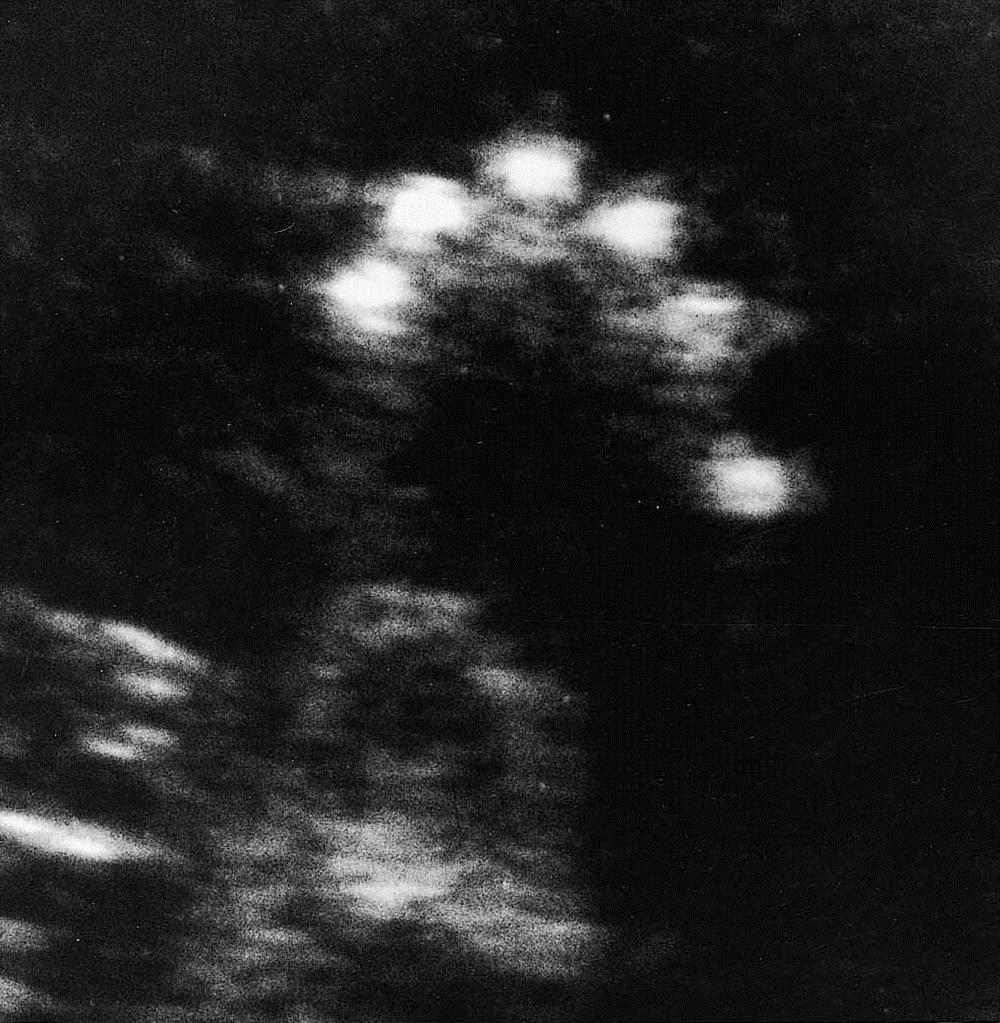
Figura 1 Imagem ultrassonográfica de corte transversal
da mão de feto evidenciando polidactilia.
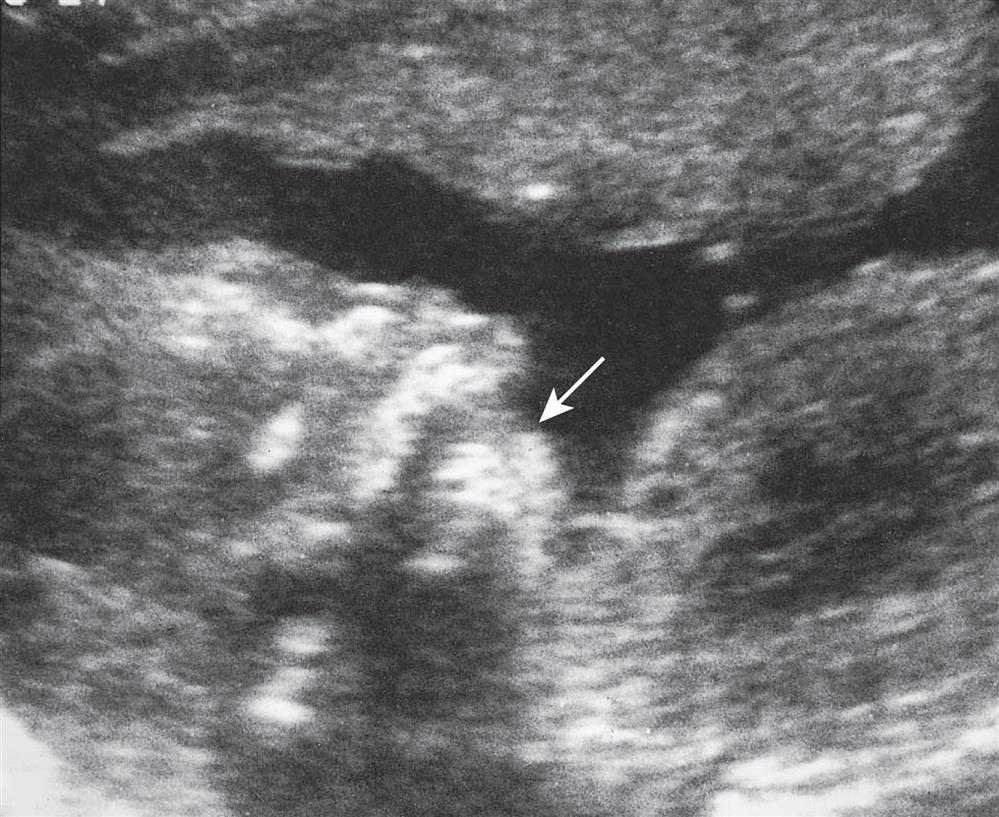
Figura 2 Imagem ultrassonográfica sagital longitudinal
da cabeça e parte superior do tórax de um feto mostrando micrognatia
(mandíbula pequena) (seta).
Atualmente, o exame de US de rotina é oferecido por volta das 12 semanas de gestação como parte da avaliação inicial da gestação, incluindo a confirmação da idade gestacional, e a visualização dos batimentos do coração. Uma visão precoce das proporções corporais fornece pistas sobre o bem-estar fetal, e um foco particular de atenção é a avaliação da espessura da coxim nucal, ou translucência nucal (TN) (Fig. 3).
O aumento da TN é observado em fetos com síndrome de Down, bem como em outras trissomias, e a medição da TN no primeiro trimestre é incorporada ao teste de triagem combinado oferecido para a síndrome de Down, Síndrome de Edwards e síndrome de Patau. Uma medida elevada de TN não é específica e pode ser observada em diversas anomalias cromossômicas, bem como em cardiopatias congênitas isoladas. O valor preditivo para trissomias tendo em vista a Translucência nucal por milimetros visualizados são respectivamente de 3mm para 13%, 4mm para 57% e 5mm para 80%.
O USS nesta fase inicial pode detectar um defeito significativo do tubo neural (DTN) e outras anomalias importantes. Depois disso, a USS é oferecida rotineiramente a todas as mulheres grávidas com cerca de 20 semanas de gestação como triagem adicional para anormalidades estruturais, porque o feto cresceu até um tamanho que torna possível a visualização detalhada.
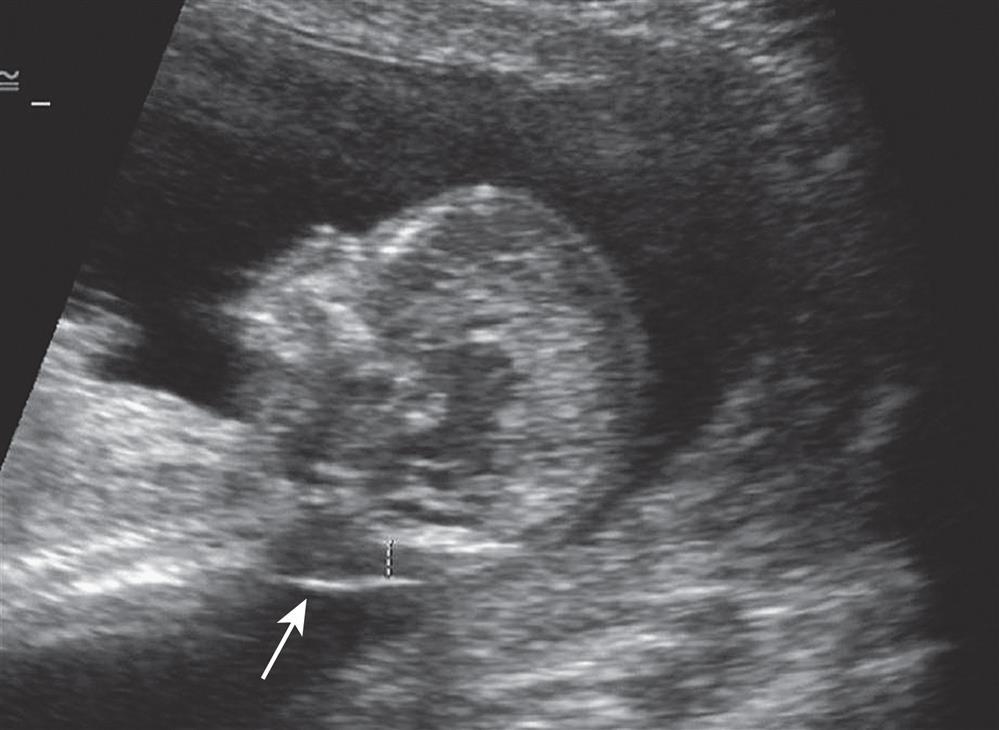
Figura 3 Espessamento da nuca – acúmulo de líquido na
parte posterior do pescoço. Quanto maior a espessura, maior a
probabilidade de haver uma anomalia cromossômica (por exemplo, síndrome
de Down) e/ou anomalia cardíaca. Essa descoberta leva à varredura
detalhada do coração fetal e, geralmente, a testes invasivos (reação em
cadeia da polimerase fluorescente quantitativa e hibridização genômica
comparativa por array).
Além das alterações aqui já mencionadas é importate atentar para as seguintes alterações mais frequentemente visualizadas no ultra-som: Hidropsia fetal não-rhesus, Hérnia diafragmática, Anencefalia, Gastrosquise, Encefalocele e Meningocele, Hipoplasia cardíaca (principalmente a Ventricular Esquerda), Lesão obstrutiva do trato urinário e a respectiva sequencia de Potter, a Agenesia do fêmur com encurtamento ipsi ou contralateral do membro.
As anomalias complexas como as apresentadas a seguir, estão mais comumente associadas a alteração de cariótipo: Hidrocefalia com Comunicação Intra Ventricular ou sinal da Dupla bolha com polidramio em feto com crescimento uterino restrito, visto em trissomia do 21, bem como Hipoplasia cardíaca (ventricular E principalmente), Onfalocele, Malformação de Dandy-Walker, Cisto de plexo coróide com dilatação da cisterna magna visto em trissomias do 18.
Os avanços na varredura fetal agora permitem imagens tridimensionais principalmente se associados à ressonância magnética (MRI), que são particularmente úteis para visualizar anormalidades do cérebro fetal. No entanto, a detecção de anomalias do cérebro em desenvolvimento pode não ser possível antes das 24 semanas de gestação. A ressonância magnética fetal a visualização com muito mais detalhes, e pode diagnosticar distúrbios graves e sutis. Na realidade, toda uma equipa de pessoas, incluindo especialistas em medicina fetal e neurorradiologistas, estaria envolvida nestas discussões e concluiria sobre o prognóstico para o feto, independentemente de um diagnóstico genético vir a ser confirmado dentro do tempo da gestação.
A cada período gestacional o ultrasom tem a capacidade progressiva de ver determinadas estruturas já desenvolvidas, a lista a seguir resume aquilo que se pode encontrar de acordo com a idade gestacional:
Até 14 semanas:
Número de embriões
Vitalidade embrio-fetal
Presença do pólo cefálico
Avaliação da coluna vertebral
Presença dos membros
17 a 20 semanas:
Avaliação do pólo cefálico
Avaliação detalhada da coluna vertebral
Avaliação da face
Presença da parede abdominal
Presença do diafragma
Visualização do estômago
Visualização da bexiga
Avaliação dos órgãos genitais externos
Avaliação detalhada dos membros e extremidades
20 a 24 semanas:
Avaliação das estruturas cerebrais
Avaliação detalhada da face
Avaliação do tórax e do coração
Avaliação detalhada do abdômen e do aparelho digestivo
Avaliação dos rins
Avaliação detalhada dos membros e extremidades
28 a 32 semanas:
Busca por anomalias esqueléticas
Investigação de nanismos
Identificação de cistos de ovário
Vale ressaltar aqui, que ao longo deste estudo abordaremos ainda mais os diagnósticos por ultrasom gestacional para que possas fixar bem as informações e distinguir as principais alterações observadas no período fetal.
Radiografia
A radiografia é uma técnica usada no passado para diagnosticar displasias esqueléticas hereditárias uma vez que o esqueleto fetal pode ser visualizado a partir da 10a semana. Ainda pode ser útil ocasionalmente, apesar da ampla disponibilidade de USS de alta resolução. No entanto os riscos associado à radiação reduzem a sua utilização de forma rotineira.
Amniocentese
Na amniocentese, 10 à 20 mL de líquido amniótico são aspirados através da parede abdominal sob orientação ultrassonográfica (Fig. 4 ), a partir da 15ª semana de gestação. A amostra é centrifugada para produzir um pellet de células e fluido sobrenadante. O fluido foi usado anteriormente para testar α-fetoproteína para diagnosticar DTNs, mas o USS substituiu esse método. O sedimento celular é ressuspenso em meio de cultura para estimular o crescimento celular. A maioria das células do líquido amniótico foi eliminada do âmnio, da pele fetal e do epitélio do trato urinário e não são viáveis, mas algumas crescerão.
Após aproximadamente 14 dias, geralmente há células suficientes para análise cromossômica, embora possa ser necessário um período mais longo antes que células suficientes sejam obtidas para ensaios bioquímicos. A análise direta de DNA usando reação em cadeia da polimerase fluorescente quantitativa (QF-PCR) é realizada, antes da conclusão das culturas celulares, como um teste rápido para aneuploidias dos cromossomos 13, 18, 21, X e Y. O ensaio usa primers marcados com fluorescência para analisar até cinco marcadores curtos de repetição em tandem de cada cromossomo após a separação do comprimento do fragmento em eletroforese em gel capilar. A quantidade de fluorescência e o tamanho dos fragmentos de DNA são quantificados, e as proporções são apresentadas graficamente (Fig. 5 ), mostrando assim quantas cópias dos cromossomos estão presentes. Este método rápido de detecção de aneuploidias comuns também pode detectar anormalidades como a triploidia bem antes do cariótipo estar pronto.
Um cariótipo completo é principalmente necessário apenas para confirmar os resultados de um QF-PCR anormal, com o teste de hibridização genômica comparativa (CGH) sendo agora o método padrão de análise cromossômica quando o teste invasivo é indicado. Se um casal optou por fazer uma amniocentese para procurar uma doença conhecida de um único gene, para a qual o feto corre risco, isso também é realizado como uma análise direta de DNA. Tanto este como o teste QF-PCR geralmente produzem resultados em 3 dias.
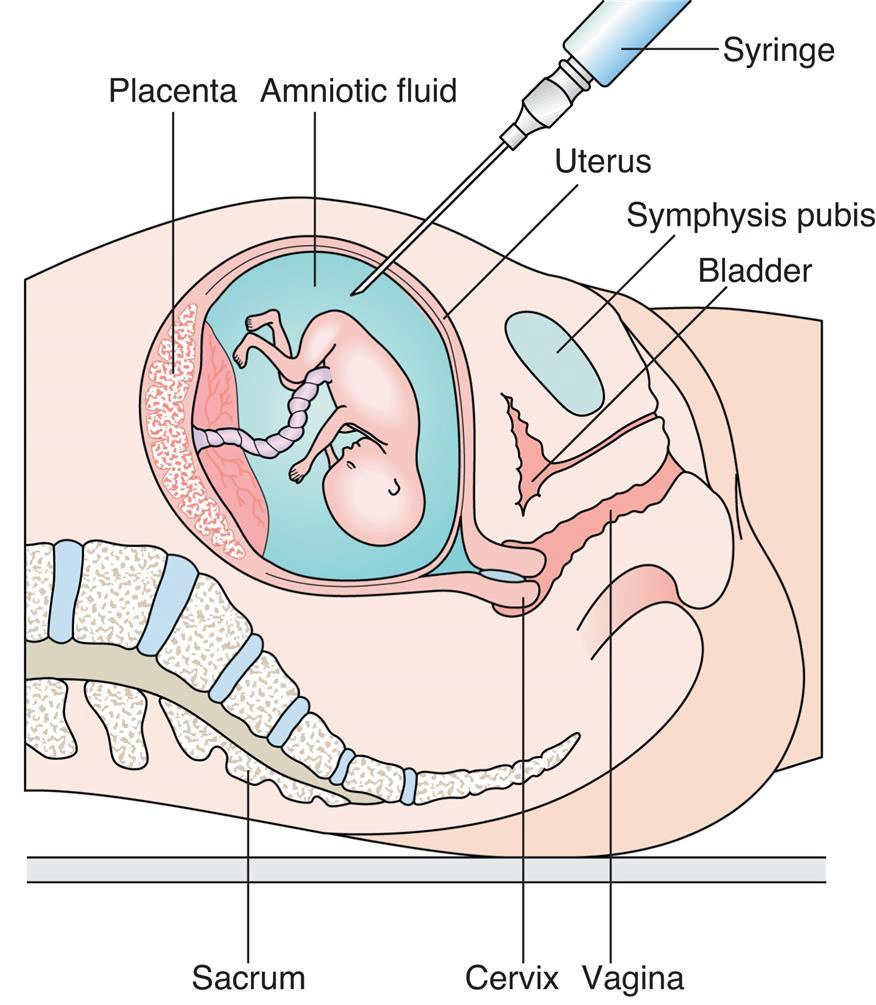
Figura 4 Diagrama da técnica de amniocentese.
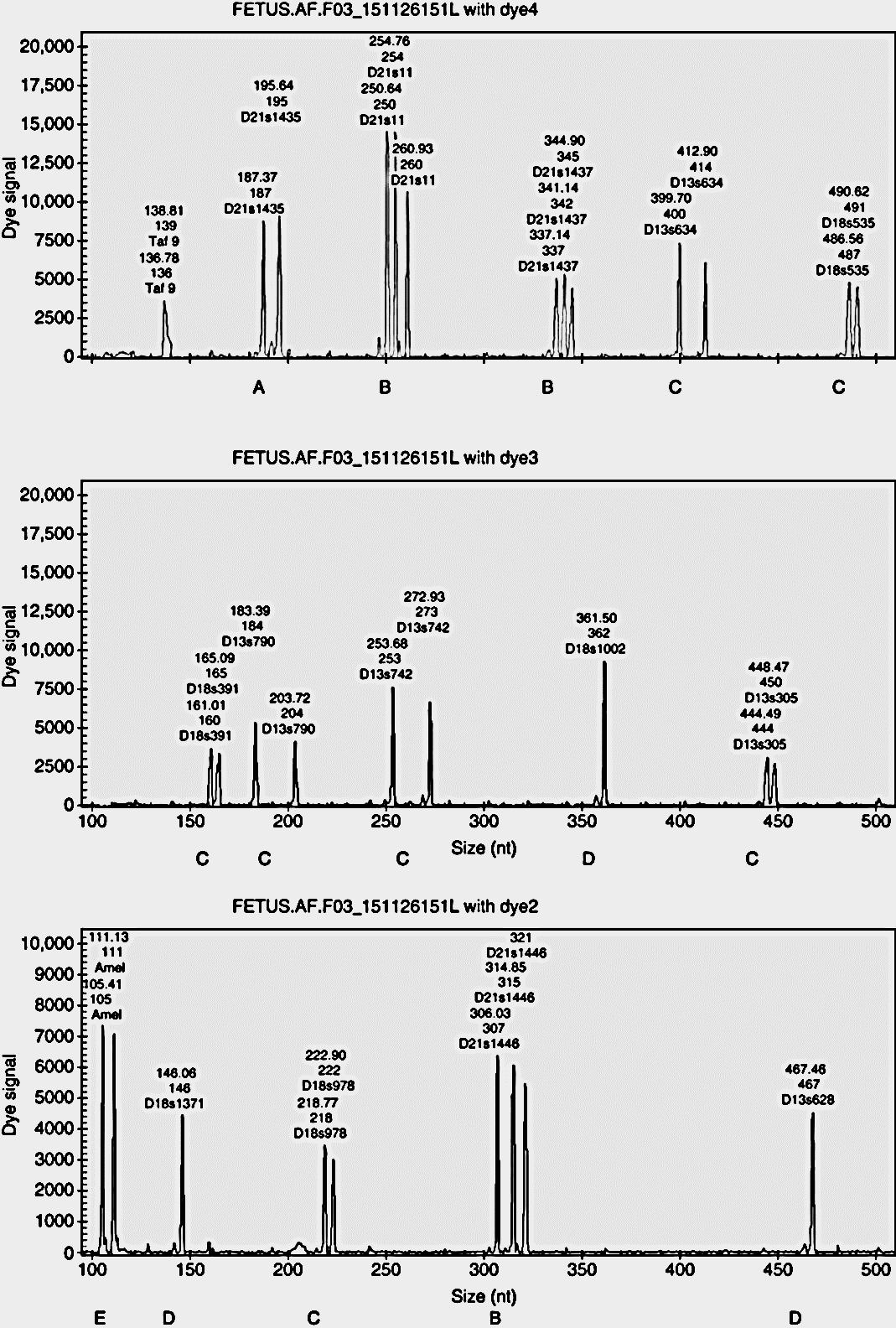
Figura 5 Resultado quantitativo da reação em cadeia da
polimerase fluorescente para um feto com síndrome de Down, trissomia 21.
(A) Um marcador bialélico para o cromossomo 21, com um pico duas vezes a
altura do outro; (B) marcadores trialélicos confirmando o diagnóstico de
trissomia do 21; (C) marcadores bialélicos para os cromossomos 13 e 18;
(D), marcadores do cromossomo 18 – picos grandes indicando duas cópias
desse cromossomo; (E) marcadores de cromossomos sexuais (em Xp22 e Yp11)
para determinar o sexo (masculino neste caso).
Quanto ao aconselhamento ao casal sobre a realização amniocentese, devemos informar do risco de 0,5% a 1% de aborto espontâneo associado ao procedimento e que, se o resultado for anormal, eles enfrentarão a possibilidade de uma interrupção da gestação no meio do trimestre, por risco de indução do parto.
Amniocentese no início da gestação, entre 12 e 14 semanas de gestação, possuem taxas de sucesso na obtenção de resultados, com um risco semelhante de aborto espontâneo. No entanto, o volume de líquido amniótico nesta fase inicial da gestação é baixo e a amniocentese precoce não é amplamente praticada. Embora fornecesse um resultado mais precoce, uma interrupção da gestação no meio do trimestre ainda é uma possibilidade se o feto for afetado.
Desta forma as principais indicações para a amniocentese para estudo citogenético são:
Idade materna acima de 35/37 anos
História familiar ou antecedente de criança com anormalidade cromossômica
História familiar ou antecedente fetal de defeito do tubo neural
Antecedente de criança com múltiplas anomalias congênitas
Anormalidade fetal (anatômica) diagnosticada pela ultra-sonografia
Principais análises realizadas na Amniocentese:
| Pesquisa | Exame praticado | Tempo de realização |
|---|---|---|
| Anomalias cromossômicas | Cariótipo fetal; sexo fetal (doenças hereditárias ligadas ao X) | A partir de 15 semanas |
Doenças metabólicas congênitas
|
Fenótipo HLA em células de cultura | A partir de 15 semanas
|
| Dosagem enzimática ou dos metabólitos em células amnióticas (diretamente ou após cultura) | ||
| Dosagem enzimática ou dos metabólitos dentro do sobrenadante do líquido amniótico | ||
Malformações do tubo digestivo
|
Dosagem de bilirrubina |
Variável
|
| Dosagem dos ácidos biliares | ||
Malformações do tubo neural
|
Dosagem de alfafetoproteína | |
| Estudo da acetilcolinesterase | ||
| Pesquisa de células de origem nervosa em cultura | ||
Análise de DNA
|
Sexo fetal |
A partir de 15 semanas
|
| Doença genética na qual o gene esteja identificado e que exista sonda molecular | ||
| Determinação de paternidade | ||
| Pesquisa de infecções por PCR (rubéola, toxoplasmose, citomegalovirus) | ||
| Mucoviscidose | Estudo das isoenzimas da fosfatase alcalina, da gamaglutamil transpeptidase, amniopeptidase M, dissacaridases | 18 semanas |
| Infecção ovular | Pesquisa bacteriológica, parasitológica, viral | Variável |
| Patologia imunológica hemolítica | Dosagem de bilirrubina em casos de anemia | Após 26 semanas |
| Maturidade pulmonar fetal | Estudo da relação lecitina/esfingomielina, pesquisa de fosfatidilglicerol e teste de Clements | Variável |
Biópsia de vilosidades coriônicas
Em contraste com a amniocentese, a amostragem de vilosidades coriônicas (CVS), permite que o diagnóstico pré-natal seja realizado durante o primeiro trimestre. Esse procedimento geralmente é realizado entre 11 e 13 + 6 semanas de gestação, sob orientação ultrassonográfica, por aspiração transcervical ou, mais comumente, transabdominal do tecido das vilosidades coriônicas (CV) (Fig. 6 ). Esse tecido é de origem fetal, sendo derivado da camada celular externa do blastocisto (isto é, o trofoblasto) e passa a formar a placenta. A decídua materna, geralmente presente na amostra da biópsia, deve ser removida antes da análise da amostra.
Biópsia placentária é o termo usado quando o procedimento é realizado em fases posteriores da gestação. A amostra das vilosidades coriônicas é separada e uma parte é montada em cultura. Do outro, o DNA é extraído para análise da doença genética para a qual o feto está em risco, ou seja, um teste de mutação direta ou, ocasionalmente, um conjunto de marcadores haplótipos de alto risco.
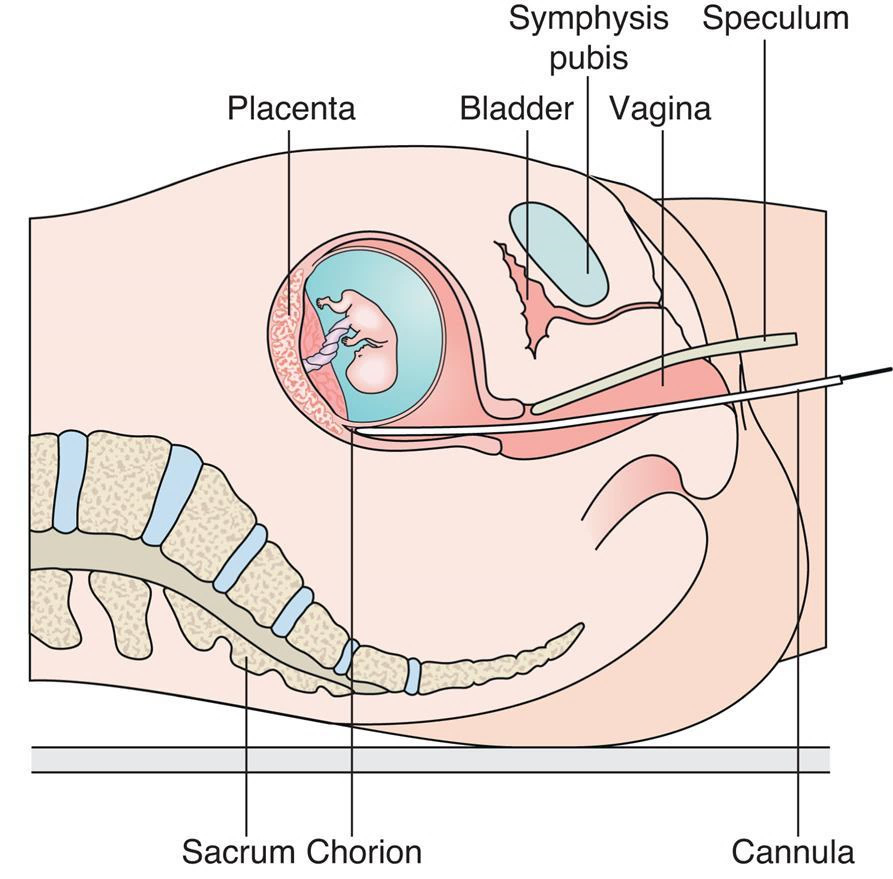
Figura 6 Diagrama da técnica de amostragem
transcervical de vilosidades coriônicas.
O QF-PCR (reação em cadeia da polimerase fluorescente quantitativa) para as aneuploidias comuns também costuma ser realizado, conforme discutido.
No entanto o Array-CGH é agora a análise cromossômica padrão para casos que atendem aos critérios de teste, e uma análise do cariótipo completa pode ser necessária após a cultura, por exemplo, para confirmar uma suspeita de translocação desequilibrada no feto, embora este não seja mais um teste padrão.
Às vezes, a análise é bioquímica (por exemplo, para erros inatos do metabolismo). Isto geralmente pode ser realizado na amostra de tecido, mas, se for muito pequeno, será realizado após a cultura.
O risco de aborto espontâneo devido ao procedimento é geralmente estimado em 1%, embora na prática, com operadores experientes, seja geralmente menor.
Principais indicações de Biópsia de vilosidades coriônicas:
Doenças Metabólicas:
Acidemia argininosuccínica
Acidemia metilmalônica
Acidemia propiônica
Adrenoleucodistrofia
Citrulinemia
Cistinose
Deficiência de piruvato carboxilase
Deficiência de adenosina deaminase
Doença de Fabry
Doença de Tay-Sachs
Doença de Sandhoff
Doença de Gaucher (I, II, III)
Doença de Gaucher tipo I
Doença de Gaucher tipo II
Doença de Gaucher tipo III
Glicogenose tipo II
Homocistinúria
Doença de Krabbe
Doença de Lesch-Nyhan
Leucinose
Leucodistrofia metacromática
Manosidose
Doença de Menkes
Mucolipidose (I, II, III)
Mucolipidose tipo I
Mucolipidose tipo II
Mucolipidose tipo III
Mucopolissacaridose:
I: Doença de Hurler
II: Doença de Hunter
III: Doença de Morquio
Mucopolissacaridose tipo I (Doença de Hurler)
Mucopolissacaridose tipo II (Doença de Hunter)
Mucopolissacaridose tipo III (Doença de Morquio)
Niemann-Pick A-B
Doença de Zellweger
Biologia Molecular:
Adrenoleucodistrofia
Alfa e Beta talassemias
Fibrose cística pulmonar
Distrofia muscular de Duchene e Becker
Hemofilia A e B
Deficiência de 21 hidroxilase
Deficiência de OTC
Fenilcetonúria
Síndrome do X frágil
Osteogênese imperfeita
Infecções Fetais (PCR):
Toxoplasmose
Rubéola
Citomegalovírus
Parvovírus B19
Herpes simples
Adenovírus
Coxsackie
Outras Indicações:
Cariótipo fetal
Idade materna acima de 35/37 anos
Translocação materna
Translocação paterna
Doença ligada ao X
Sinais precoces de alterações ultra-sonográficas
Fetoscopia
A fetoscopia envolve a visualização do feto por meio de um endoscópio. Em grande medida, esta técnica foi substituída pela USS detalhada, outras técnicas de imagem e testes genéticos para obter um diagnóstico.
A fetoscopia é usada quando intervenções cirúrgicas no bebê em desenvolvimento podem prevenir danos irreversíveis, por exemplo, a inserção de um dreno no trato urinário para prevenir danos secundários das válvulas uretrais posteriores e o tratamento de bandas amnióticas e síndrome de transfusão entre gêmeos. Poderia ser usado para coletar amostras específicas de biópsia para auxiliar no diagnóstico; no entanto, o procedimento apresenta risco significativo de perda fetal ou parto prematuro.
Portanto, qualquer intervenção deste tipo deve ser cuidadosamente considerada em termos dos seus riscos e benefícios tanto para o bebé como para a mãe. Quaisquer procedimentos desse tipo seriam realizados apenas em centros especializados.
As principais indicações para Biopsias e punções fetais são:
Biópsia de pele
Alterações na pigmentação
Epidermólise bolhosa
Alteração na queratinização
Displasia ectodérmica anidrótica
Biópsia hepática
Alterações enzimáticas ligadas ao cromossomo X (fetos masculinos)
Glicogenose
Déficit enzimático do ciclo da uréia
Fenilcetonúria
Punção vesical e/ou renal
Para diagnóstico de função renal em uropatia obstrutiva
Punção de derrames serosos e formações císticas
Para diagnóstico citológico, bioquímico ou enzimático
Ascite
Derrame pleural e pericárdico
Hidropsia
Cisto pulmonar
Cisto renal
Cordocentese
A fetoscopia já foi um procedimento usado para obter uma pequena amostra de sangue fetal de um dos vasos do cordão umbilical no procedimento conhecido como cordocentese, mas isso raramente é necessário com a visualização agora fornecida pelo moderno UltraSom.
A coleta de sangue fetal é possível a partir de cerca de 20 semanas de gestação e é usada rotineiramente no tratamento da isoimunização contra Rhesus (RH+), bem como em alguns casos de hidropsia fetal não imune em que há suspeita de hemoglobinopatia. Ocasionalmente, uma amostra para análise cromossômica pode ajudar a resolver problemas associados a um possível mosaicismo em amostras de vilosidades coriônicas ou amniocentese.
As principais indicações da Cordocentese são:
Diagnóstico pré-natal
Cariótipo rápido
Idade gestacional avançada
Dúvidas no exame de LA
Malformação no exame de ultra-som
Síndrome X frágil
Doenças genéticas
Distúrbios da coagulação dos glóbulos vermelhos
Metabólicas
Imunológicas
Infecções congênitas
Toxoplasmose
Rubéola
CMV, varicela
Outras
Aloimunizações
Fator Rh
Plaquetas
Controle do bem-estar fetal
Púrpura trombocitopênica idiopática
Restrição de crescimento intra-uterino
Hidropisia não-imune
Outras
Para fins de Terapia fetal:
Transfusões intra-uterinas
De Eritrocitos
De Plaquetas
Aplicação de Drogas
Curare
Digoxina
Outras
Seguimento da terapia materna com dosagem de:
Corticoides
IgG
Antibióticos
Oxigênio
Observações:
Esta lista não é exaustiva e outras indicações podem existir.
A decisão de realizar a cordocentese deve ser tomada em conjunto com o médico obstetra e geneticista.
Triagem pré-natal
A história do rastreio pré-natal acessível e generalizado, começou no início da década de 1970, com a descoberta de uma associação entre níveis elevados de α-fetoproteína (AFP) no soro materno e DTN. A estimativa dos níveis de AFP foi gradualmente introduzida nos serviços clínicos, e desde então, a triagem pré-natal tem evoluido progressivamente.
Com o avanço do rastreio bioquímico e molecular, atualmente é possível realizar de forma privada o rastreio de portadores, incluindo os “panétnicos”, que abrange mais de 2000 variantes comuns observadas em 250 doenças, em grande parte de herança autossómica recessiva ou recessiva ligada ao X. Uma vasta gama de doenças relativamente raras pode ser rastreada com base no facto de serem mais comuns em grupos populacionais específicos, inclusive aqueles grupos originalmente isolados com endogamia múltipla e, portanto, com certas variantes patogénicas prevalentes.
Também está disponível exames específico para condições comuns observadas na população judaica Ashkenazi, que inclusive consta com o financiamento por uma instituição de caridade chamada Jnetics; entre elas as condições estão a Doença de Tay-Sachs, fibrose cística, disautonomia familiar, doença de Canavan, distúrbio de armazenamento de glicogênio tipo 1a, anemia de Fanconi, doença de Niemann-Pick tipo A, síndrome de Bloom e a mucolipidose IV. Além destas temos a ataxia telangiectasia (em judeus norte-africanos), distrofia muscular de cinturas (em judeus líbios) e síndrome de Costeff (em judeus iraquianos).
A grande adesão dos casais e a evolução dos rastreios pré-natais faz com que atualmente métodos não invasivos como a pesquisa de DNA fetal livre de células da circulação materna seja cada vez mais aprimorado.
Triagem em soro materno
A triagem sérica materna é oferecida desde a década de 1980 para síndrome de Down (trissomia 21), síndrome de Edwards (trissomia 18) e síndrome de Patau (trissomia 13) usando uma amostra de sangue obtida da mãe por volta das 12 semanas de gestação. Isto é combinado com a medição da Transluscência Nucal no exame do primeiro trimestre e na idade materna, para estabelecer um risco combinado para cada trissomia.
Este método de triagem detecta cerca de 90% dos casos de síndrome de Down, com taxas de detecção ligeiramente mais altas para trissomia 13 e 18.
Nos casos em que o rastreio do primeiro trimestre não foi possível, é oferecido às mulheres o rastreio quádruplo entre as 14 e as 20 semanas de gestação. Isso rastreia apenas a síndrome de Down e é menos preciso do que o risco combinado do primeiro trimestre. Falaremos mais adiante sobre estes testes.
Defeitos do tubo neural
Foi observado no início da década de 70, que muitas gestações em que o bebé tinha uma Defeito de Tubo Neural aberto, podiam ser detectadas pela 16a semana de gestação através da análise de a-fetoroteína no soro materno. AFP é o “equivalente” fetal da albumina e é a principal proteína do sangue fetal. Se o feto tiver um DTN aberto, o nível de AFP aumenta tanto no líquido amniótico quanto no soro materno como resultado do vazamento do defeito. Os DTN abertas são condições graves, incluíndo a anencefalia, e entre 80% a 90% da pequena proporção de bebés que sobrevivem com uma lesão lombossacra aberta ficam majoritariamente prejudicados.
Infelizmente, o rastreio de AFP no soro materno para DTN não é 100% sensível nem 100% específico. As curvas para os níveis de AFP sérica materna em gestações normais e afetadas se sobrepõem (Fig. 7 ), de modo que, é necessário introduzir um nível de corte arbitrário, abaixo do qual nenhuma ação adicional é tomada. Geralmente é o percentil 95 ou 2,5 múltiplos da mediana; Desta forma, cerca de 75% dos casos de espinha bífida aberta são detectados. Como a maioria das anomalia fetais são visualizadas por US de 20 semanas, isso geralmente é suficiente para diagnosticar DTNs, desde que o ultrassonografista consiga obter boa visualização do sistema nervoso.
O escaneamento com US substituiu majoritariamenete a triagem sérica materna para DTN. Com o US é possível observar a anencefalia como uma deficiência nas crânio (Fig. 8 ), e uma mielomeningocele aberta está quase invariavelmente associada à herniação das tonsilas cerebelares através do forame magno. Isso deforma os hemisférios cerebelares, que passam a ter uma aparência curva conhecida como “sinal da banana”; a testa também fica distorcida, dando origem a um formato conhecido como “sinal do limão” (Fig. 9 ).
Uma encefalocele posterior é facilmente visualizada como um saco na região occipital (Fig. 10 ) e sempre estimula a busca por anomalias adicionais que possam ajudar a diagnosticar uma condição reconhecível, por exemplo, a síndrome de Meckel-Gruber.
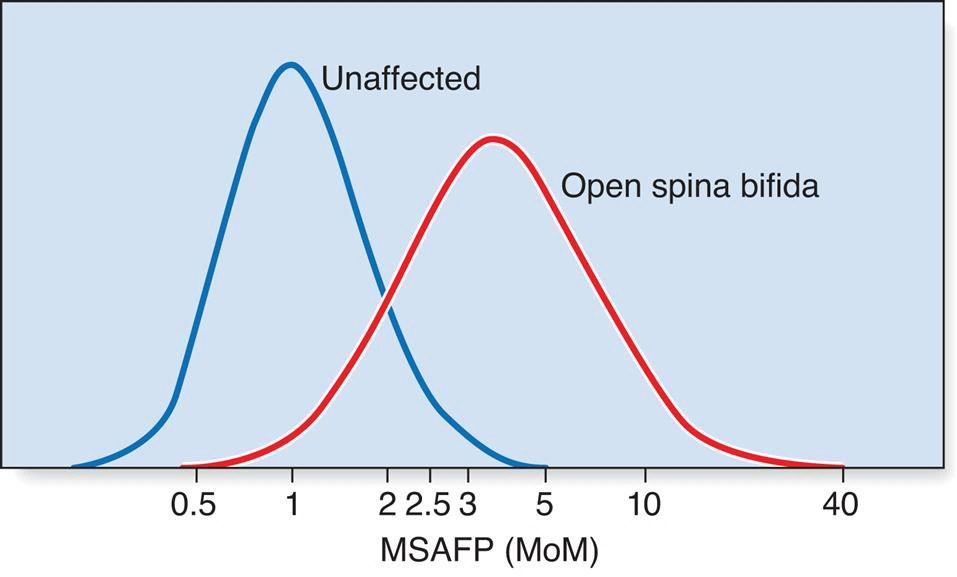
Figura 7 Níveis de α-fetoproteína sérica materna
(MSAFP) às 16 semanas de gestação plotados em uma escala
logarítmica como múltiplos da mediana (MoMs). Mulheres com um
valor de 2,5 MoMs ou superior recebem investigações adicionais.
Modificado de Brock DJH, Rodeck CH, Ferguson-Smith MA, eds.
Diagnóstico e triagem pré-natal . Edimburgo: Churchill
Livingstone; 1992.
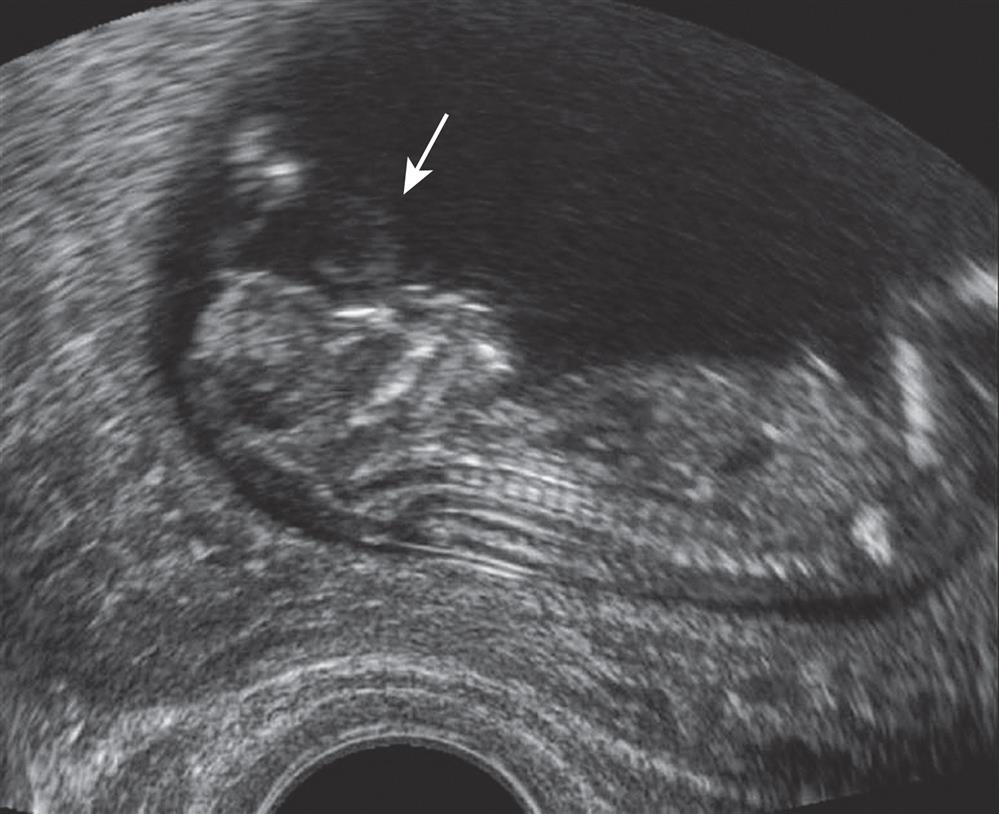
Figura 8 Anencefalia (seta). Não há crânio e
esta forma de defeito do tubo neural.
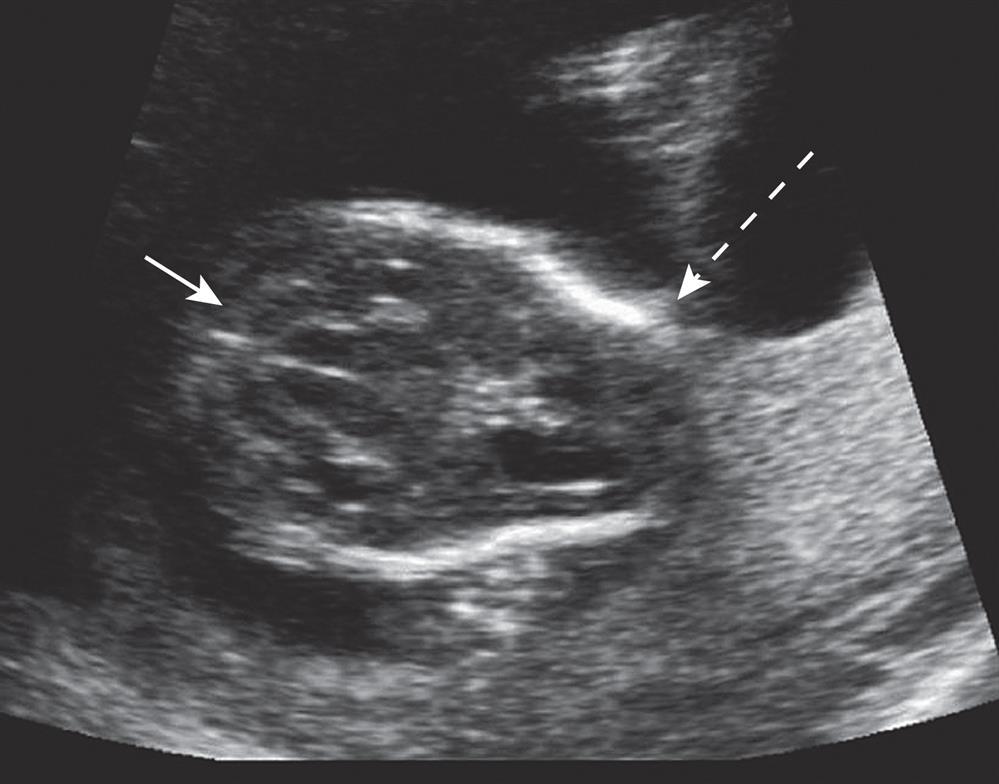
Figura 9 O chamado sinal da banana mostrando a distorção dos hemisférios cerebelares em uma estrutura curva (seta sólida). A testa também está distorcida em um formato conhecido como “sinal do limão” (seta quebrada).
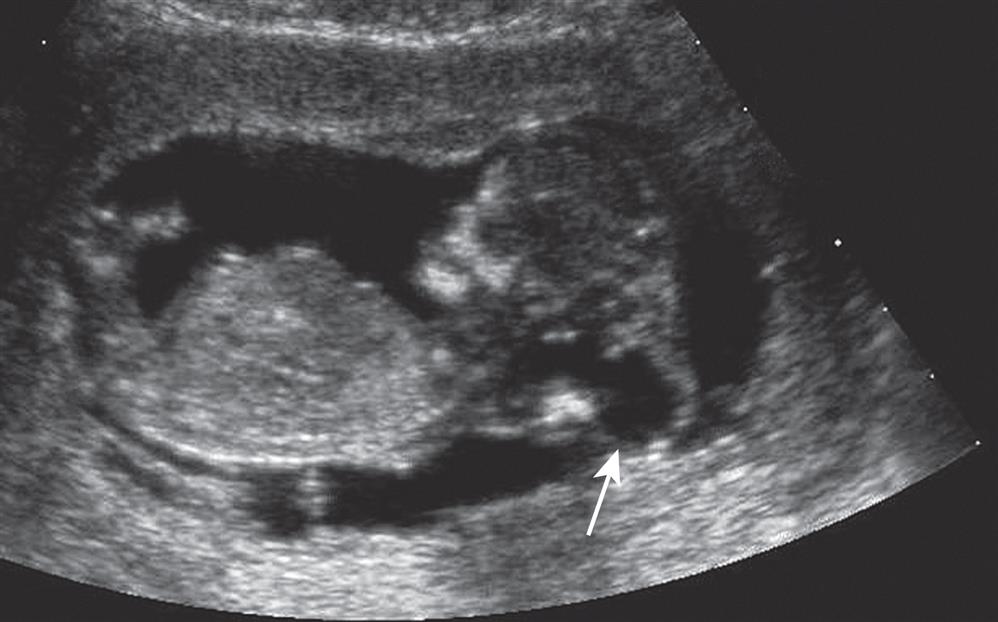
Figura 10 Encefalocele posterior (seta), uma forma rara de defeito do tubo neural. Este pode ser um achado isolado ou associado à polidactilia e alterações renais císticas na síndrome de Meckel-Gruber.
Uma concentração elevada de AFP no soro materno não é específica para DTN abertas (Quadro 1 ). Outras causas incluem ameaça de aborto espontâneo, gestação gemelar e anomalia fetal, como exônfalo, em que há protrusão do conteúdo abdominal através do umbigo.
Causas do aumento do nível de Α-fetoproteína no soro materno
Anencefalia
Espinha bífida aberta
Idade gestacional incorreta
Sangramento fetal intrauterino
Ameaça de aborto
Gestação múltipla
Síndrome nefrótica congênita
Defeito na parede abdominal
Como resultado destas modalidades de rastreio, a incidência de nascimentos de DTN abertas, caiu drasticamente. Outros factores que contribuíram foram uma melhoria geral na dieta e a introdução da suplementação periconcepcional de ácido fólico.
Síndrome de Down e outras anomalias cromossômicas
O teste combinado
A confirmação de uma anomalia cromossômica no feto requer estudos citogenéticos ou moleculares usando material obtido por um procedimento invasivo como BVC ou amniocentese. Contudo, as anomalias cromossómicas, em particular a síndrome de Down, a síndrome de Edwards e a síndrome de Patau, podem ser rastreadas durante a gestação tendo em conta factores de risco como a idade materna, os níveis de marcadores bioquímicos no soro materno (Tabela 2 ) e a TN.
Tabela 2. Fatores de risco maternos para síndrome de Down
| Idade Avançada (≥35 anos) / Soro Materno | Valores Múltiplos da Mediana * |
| α-fetoproteína | (0,75) |
| Estriol não conjugado | (0,73) |
| Gonadotrofina coriónica humana | (2.05) |
| Inibina-A | (2.10) |
* Os valores entre parênteses referem-se aos valores médios nas gestações afetadas, expressos como múltiplos da mediana (MoMs) nas gestações normais.
O uso de marcadores bioquímicos na triagem pré-natal baseou-se na descoberta de que, às 16 semanas de gestação, os níveis séricos maternos de AFP e estriol não conjugado tendiam a ser mais baixos em gestações com síndrome de Down em comparação com o normal, enquanto o nível sérico materno de gonadotrofina coriônica humana (hCG) geralmente estava elevado. Outras pesquisas confirmaram marcadores bioquímicos capazes de contribuir para a quantificação do risco no primeiro trimestre, o que por sua vez levou ao rastreio combinado amplamente utilizado atualmente.
O teste combinado do primeiro trimestre mede os níveis de beta-hCG e proteína plasmática A associada à gestação (PAPP-A) no soro materno. A PAPP-A é produzida pela placenta e acredita-se que regule múltiplos fatores responsáveis pelo crescimento placentário. Níveis baixos no primeiro trimestre estão associados às três trissomias. Os resultados bioquímicos são combinados com a idade materna e a idade gestacional (com base no comprimento cabeça-nádega) para calcular a probabilidade geral de que o feto seja afetado pela trissomia 21, 18 ou 13. Isso é expresso como uma probabilidade, com qualquer risco excedendo 1 em 150 recebem testes adicionais. Isto pode ser na forma de BVC, mas em alguns centros seriam agora oferecidos testes pré-natais não invasivos (NIPT). Esta técnica, que utiliza DNA fetal livre na circulação materna, não é um teste diagnóstico; no entanto, se o risco for baixo no NIPT, podem ser evitados testes mais invasivos.
Observamos que o teste combinado teve uma taxa de detecção de 90% ou mais para trissomias 21, 18 e 13, com uma taxa de falsos positivos de 4%. Esta foi a taxa de detecção num nível de risco de 1 em 150 a termo, ou quando testes invasivos são recomendados. Além disso, o teste também identificou quase todos os casos de monossomia X (síndrome de Turner) e triploidia, bem como mais de 50% de outras anomalias cromossômicas. A incorporação da medição da frequência cardíaca fetal no algoritmo de risco combinado melhorou a taxa de detecção da síndrome de Down, embora não tenha tido efeito na detecção de outras trissomias e não seja uma parte padrão deste teste.
O NIPT tem potencial adicional para melhorar o rastreio no primeiro trimestre. O NIPT demonstrou taxas de detecção com valor preditivo positivo mais alto para trissomia 21 (99%), trissomia 18 (96%) e trissomia 13 (91%), com uma taxa de falsos positivos muito menor de 0,35%.
A triagem universal com NIPT melhoraria as taxas de detecção destas três trissomias; no entanto, a sua implementação seria dispendiosa se realizado de forma rotineira em todas as gestações com baixo custo-benefícios se aplicado aos programas públicos de rastreio pré-natal, já que a capacidade de detectar outras anomalias cromossómicas e defeitos fetais importantes é variável, por exemplo, o VPP para trissomias do 7 ou 16 são bastante baixas.
Uma solução mais adequada seria combinar os dois testes de rastreio, como mencionado acima, reservando a utilização do NIPT para aqueles com determinados níveis de risco. Ao fazê-lo, o rastreio tornar-se-á cada vez mais preciso e a necessidade de testes pré-natais invasivos continuará a diminuir.
O Teste Quádruplo
Para aquelas onde a medição da TN não é possível no primeiro trimestre, é oferecida a triagem quádrupla. Esta técnica combina quatro marcadores bioquímicos: AFP, hCG, estriol não conjugado (uE3) e inibina-A, para produzir uma avaliação de risco apenas para trissomia 21. Níveis baixos de uE3 e níveis elevados de inibina-A estão associados à síndrome de Down no segundo trimestre. Tal como acontece com o teste combinado, o resultado é combinado com a idade materna, e é necessária a datação da gestação, desta vez pela medição do perímetro cefálico fetal, em vez do comprimento cabeça-nádega. Se a cabeça fetal medir 101 mm ou mais, o teste pode ser oferecido. Este método de rastreio tem uma taxa de detecção de trissomia 21 mais baixa e uma taxa mais elevada de resultados de rastreio positivos do que o teste combinado, mas é o teste de rastreio recomendado, se necessário, no segundo trimestre.
Ultrassonografia
Como já mencionado anteriormente, o ultrasom deve ser realizado por volta da 12ª semana de gestação para avaliação, entre outras coisas, da TN fetal (ver Fig. 3 ). Como já tido, o aumento do TN associa-se com um risco de trissomia autossômica (trissomias do 21, 13 e 18;), bem como a síndrome de Turner e triploidias, e outras anomalias fetais e síndromes raras.
O risco de síndrome de Down correlaciona-se com valores absolutos de TN aumentados, bem como com a idade materna (Fig. 11 ), mas, como a TN também aumenta com a idade gestacional, deve-se relacionar o risco ao valor do percentil para a idade gestacional específica. Cerca de 80% dos fetos com síndrome de Down apresentam TN acima do percentil 95. Alguns bebês com síndrome de Down apresentam atresia duodenal, que aparece como um “sinal de bolha dupla” na USS posterior do abdome fetal (Fig. 12 ).
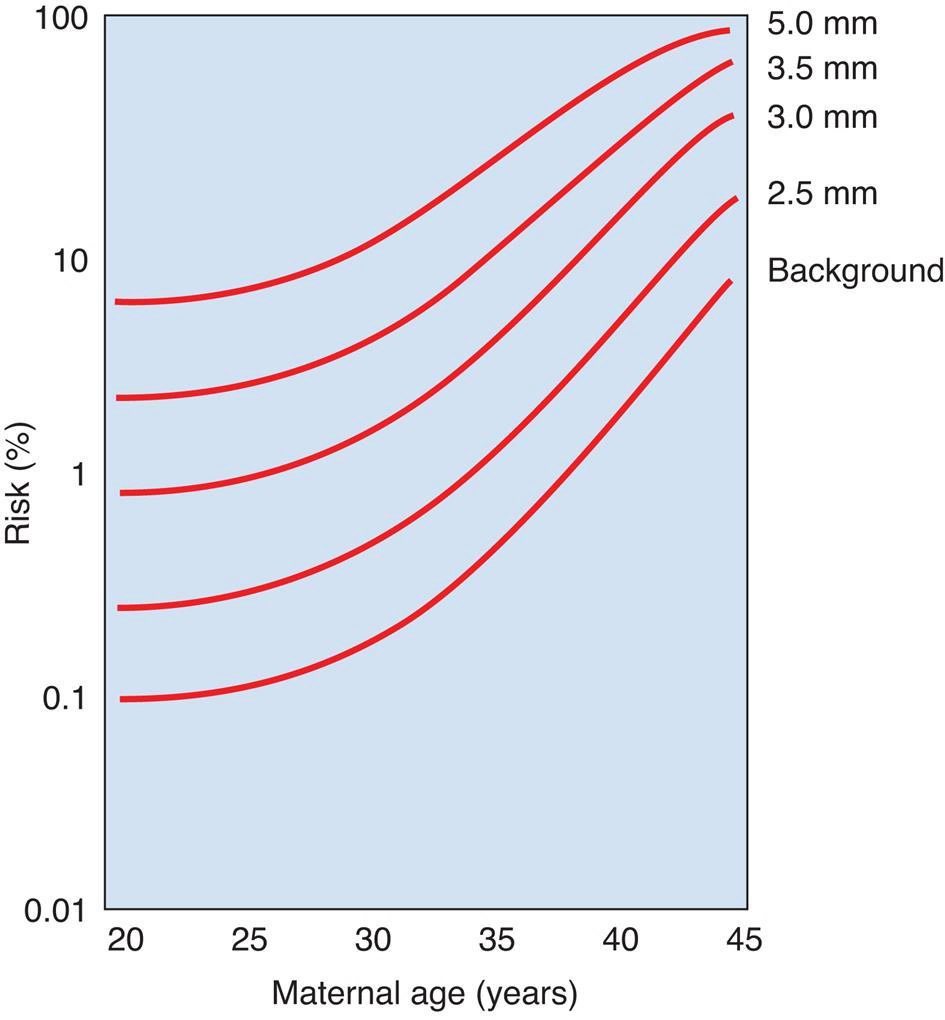
Figura 11 Risco de trissomia do 21 (síndrome de Down) por idade materna, para diferentes valores absolutos de translucência nucal com 12 semanas de gestação.
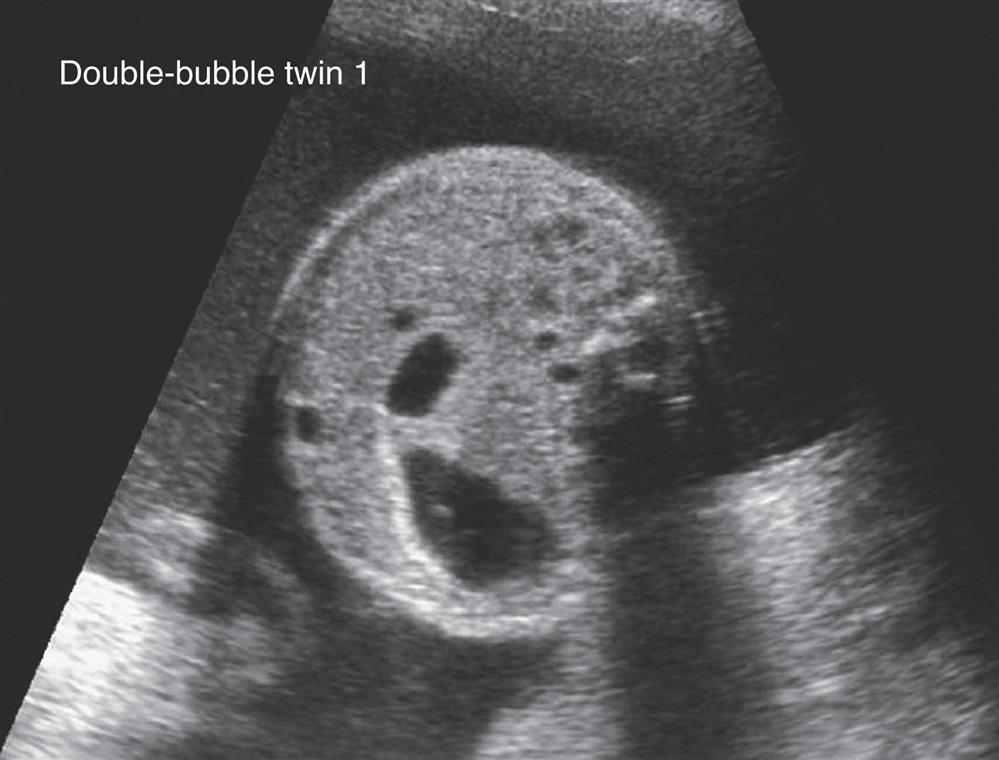
Figura 12 O “sinal da bolha dupla”, sugestivo de atresia duodenal, às vezes associado à síndrome de Down.
A cintilografia de anomalias fetais, pode ser realizada por volta das 20 semanas de gestação, e é capaz de levantar a suspeita de anomalias cromossômicas, por exemplo, se for observado exonfalos (Fig. 13 ), ou pé em cadeira de balanço (Fig. 14 ) (Tabela 3 ). Uma anormalidade cromossômica é encontrada em 50% dos fetos com exonfalos identificados às 18 semanas, e um pé em forma de balanço é característico, embora não específico, da trissomia do 18, na qual o retardo de crescimento é invariável.
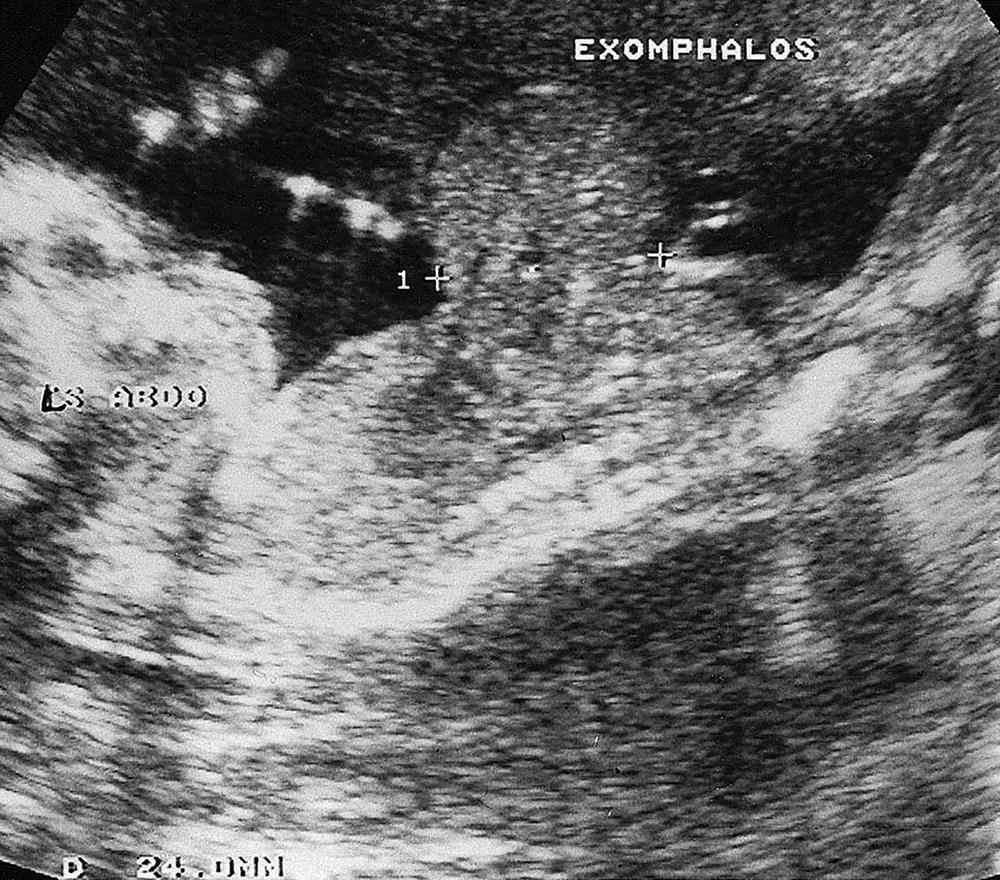
Figura 13 Ultrassonografia com 18 semanas mostrando exonfalos.
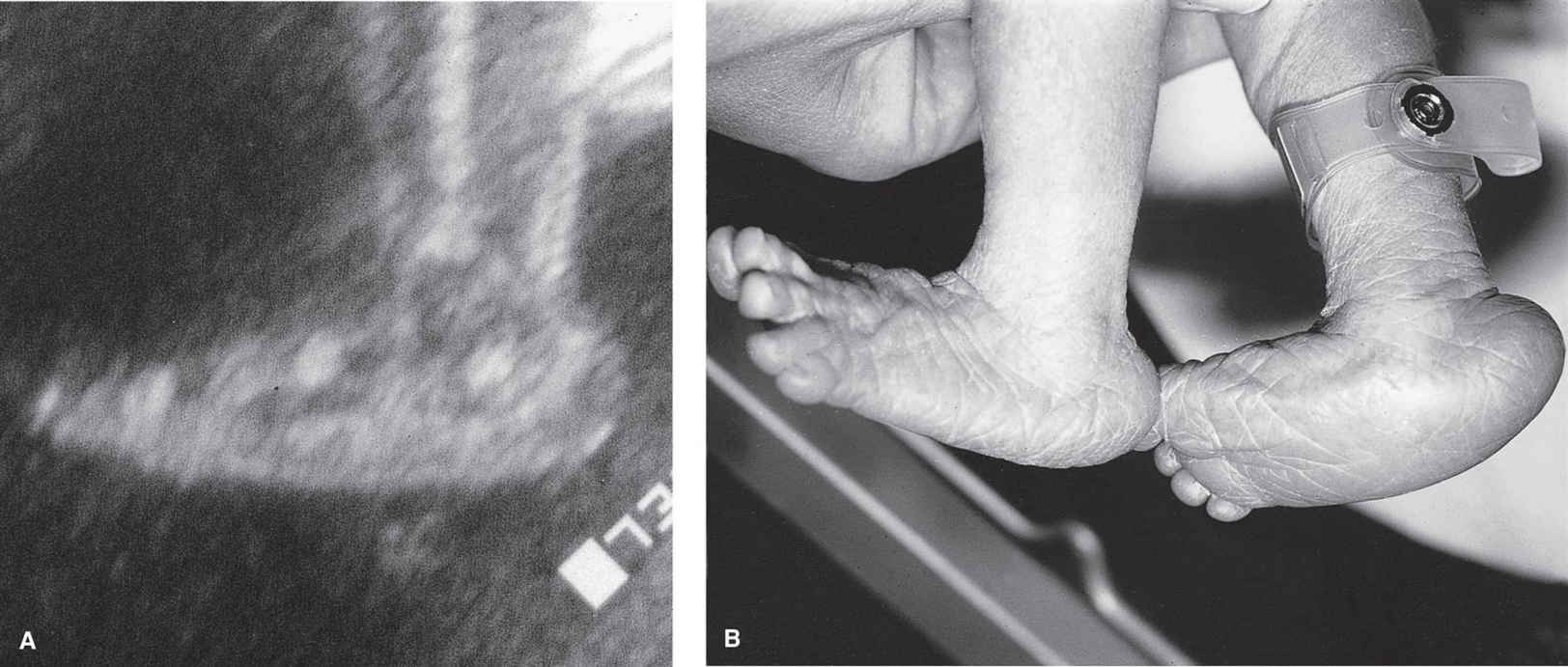
Figura 14 (A) Ultrassonografia com 18 semanas mostrando pé em balanço em um feto posteriormente diagnosticado com trissomia 18. (B) Fotografia dos pés de um recém-nascido com trissomia 18.
Tabela 3. Achados ultrassonográficos pré-natais sugestivos de anomalia cromossômica
| Alteração | Anormalidade cromossômica |
| Defeito cardíaco (especialmente canal atrioventricular comum) | Trissomia 13, 18, 21 |
| Dedos sobrepostos cerrados | Trissomia 18 |
| Higroma cístico ou hidropisia fetal | Trissomia 13, 18, 21 |
| Atresia duodenal | 45,× (síndrome de Turner) Trissomia 21 |
| Exômfalos | Trissomia 13, 18 |
| Pé com planta arqueada com calcâneo proeminente | Trissomia 18 |
Indicações para testes pré-natais
Aos casais com risco anterior elevado ou aumentado de ter um bebé com uma doença genética grave são normalmente oferecidos testes pré-natais e, idealmente, devem apresentar-se e ser avaliados antes de iniciarem uma gestação para permitir aconselhamento e tomada de decisão sem pressa. Certas comunidades judaicas ortodoxas estão extremamente bem organizadas a este respeito em relação à doença de Tay-Sachs. Na vida real, muitas vezes, muitos casais em risco aumentado devido à sua história familiar mais ampla, ou à sua própria história reprodutiva anterior, não se manifestam, ou não são encaminhados, até que a gestação esteja em andamento. Em alguns casos, pode ser demasiado tarde para realizar uma investigação clínica e laboratorial mais completa em preparação para o diagnóstico pré-natal.
Idade Materna Avançada
Esta tem sido uma indicação comum para a triagem pré-natal devido à associação entre o avanço da idade materna e o risco gestacional de trissomias autossómicas. Contudo, devido a elevada taxa de detecção do rastreio pré-natal, a idade materna avançada (>35 anos) isoladamente, não é uma indicação para testes invasivos, embora ainda seja um critério aceite em alguns países. Curiosamente, apesar dos avanços no rastreio, o número absoluto de nascimentos com síndrome de Down mudou muito pouco durante o período de 1990 a 2010, embora o número de diagnósticos pré-natais feitos tenha aumentado, o que é atribuído à idade ligeiramente mais avançada em que as mulheres têm agora filhos. Também pode haver uma vontade crescente de criar um filho com a doença, e os indivíduos com síndrome de Down vivem mais tempo.
| Incidência da Idade Materna no Parto (Anos) Síndrome de Down | |
|---|---|
| 20 | 1 em 1500 |
| 25 | 1 em 1350 |
| 30 | 1 em 900 |
| 35 | 1 em 400 |
| 36 | 1 em 300 |
| 37 | 1 em 250 |
| 38 | 1 em 200 |
| 39 | 1 em 150 |
| 40 | 1 em 100 |
| 41 | 1 em 85 |
| 42 | 1 em 65 |
| 43 | 1 em 50 |
| 44 | 1 em 40 |
| 45 | 1 em 30 |
| Adaptado de Cuckle HS, Wald NJ, Thompson SG 1987.: Estimativa do risco de uma mulher ter uma gestação associada à síndrome de Down usando sua idade e nível sérico de alfa-fetoproteína. Ir J Obstet Gynaecol 94:387–402. | |
Incidência aproximada de trissomia 21 no momento da amostragem de vilosidades coriônicas (CVS) (11–12 semanas), amniocentese (16 semanas) e parto (Term).
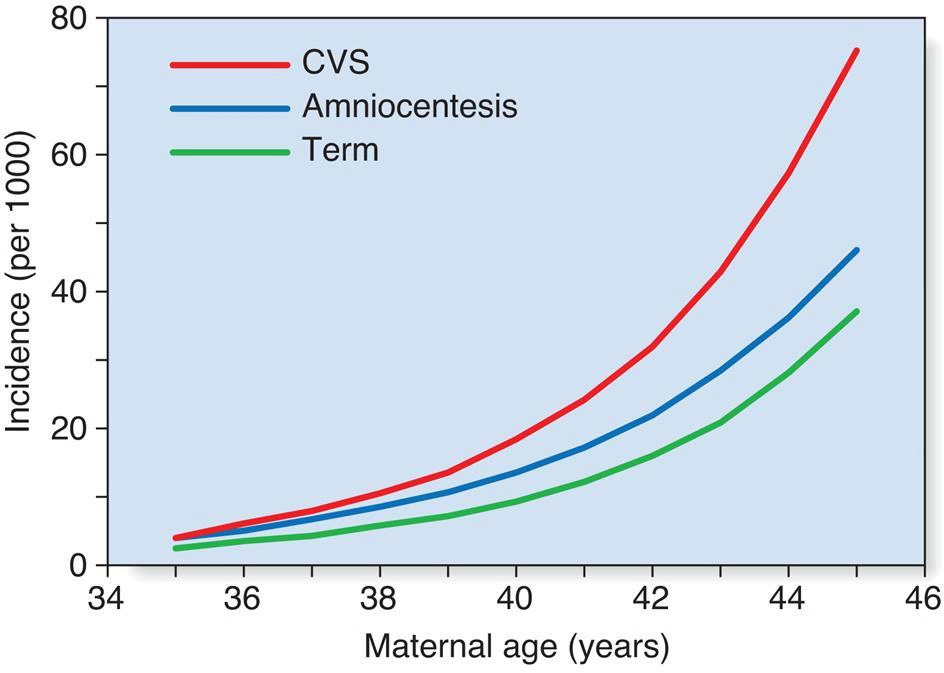
Taxas específicas da idade materna de 47, 121 e outras anormalidades citogenéticas diagnosticadas no primeiro trimestre de gestação em amostras de biópsia de vilosidades coriônicas. Am J Hum Genet 42:797 –807; e Cuckle HS, Wald NJ, Thompson SG 1987 Estimando o risco de uma mulher ter uma gestação associada à síndrome de Down usando sua idade e nível sérico de alfa-fetoproteína. Br J Obstet Gynaecol 94:387–402. Dados de Hook EB, Cross PK, Jackson L, et al 1988).
Criança anterior com anomalia cromossômica
Embora haja uma série de séries com números de risco de recorrência ligeiramente diferentes, para casais que tiveram um filho com síndrome de Down devido à não disjunção ou a uma translocação Robertsoniana desequilibrada de novo , o risco em uma gestação subsequente é geralmente dado como relacionado à idade da mãe, com risco a mais aproximadamente de 1%.
Se for descoberto que um dos pais apresenta um rearranjo cromossômico equilibrado, como uma translocação cromossômica ou inversão pericêntrica , isso fez com que um filho anterior nascesse com problemas sérios por causa de uma anomalia cromossômica desequilibrada, o risco de recorrência provavelmente estará entre 1% - 2% e 15% - 20%. O risco preciso depende da natureza do rearranjo dos cromossomos parentais e dos segmentos específicos dos cromossomos individuais envolvidos.
História familiar de uma anomalia cromossômica
Os casais podem ser encaminhados devido a um histórico familiar de anomalia cromossômica, por exemplo, síndrome de Down no filho de um irmão ou um rearranjo cromossômico equilibrado em um irmão. Como a maioria dos casos de trissomia 21 terá surgido como resultado de não disjunção, e não como resultado de uma translocação familiar, a maioria dos casais não teria risco maior do que a população em geral, e testes pré-natais invasivos não seriam indicados. Contudo, cada situação deve ser avaliada cuidadosamente, quer através da confirmação da natureza da anomalia cromossómica no indivíduo afectado, quer, se tal não for possível, através de uma análise cromossómica urgente do progenitor em risco. Quando existe um rearranjo cromossómico equilibrado conhecido num membro da família, a análise é facilmente oferecida a outras pessoas em risco, o que pode necessitar de ser providenciado com urgência caso uma gestação já esteja em curso. Se for descoberto que um dos pais apresenta um rearranjo, testes invasivos na gestação podem ser oferecidos.
História familiar de mutações patogênicas de gene único
Se os futuros pais já tiveram um filho afetado, ou se um dos pais for afetado ou tiver um histórico familiar positivo de uma doença monogênica que represente um risco significativo para a prole, então a opção de testes pré-natais deve ser discutida com eles. Apesar dos testes pré-natais apresentarem risco muito pequeno de aborto espontâneo, a maioria dos casais não escolhem este caminho. Portanto, os testes para doenças de um único gene na gestação seriam geralmente realizados apenas para doenças genéticas com consequências graves ou que limitam a vida.
História familiar ou filho anterior com anomalias estruturais congênitas
De acordo com a prática genética clínica padrão, um heredograma familiar é fundamental pois permite uma avaliação de risco empírico. Se o risco de gestação aumentar e nenhum teste genético puder ser oferecido, a USS fetal detalhada pode ser oferecida a partir da 16ª semana de gestação. Esta abordagem aplicar-se a casais que tiveram um filho com qualquer malformação ou anomalia onde os testes genéticos não identificaram um diagnóstico, mas onde se considera provável um risco de recorrência. Mesmo com testes extensivos, muitas vezes incluindo sequenciamento do exoma, nem sempre é possível chegar a um diagnóstico. Nesse cenário, exames detalhados, eco fetal ou ressonância magnética cerebral podem ser usados para procurar sinais de recorrência.
História familiar de dificuldade de aprendizagem não diagnosticada
Casais que em vigência de uma gestação já tem um filho, ou parente próximo, com dificuldade de aprendizagem não diagnosticada, com ou sem características dismórfica se beneficiam da análise por CGH-array do caso índice (Figura 5.7 ) e testes de síndrome do X-frágil, se apropriado. Cada vez mais, a tecnologia de sequenciamento de próxima geração será usada neste cenário onde o teste CGH-array é normal e há suspeita de uma causa de dificuldade de aprendizagem de um único gene. Quando o casal já tem um filho com graves dificuldades de aprendizagem, por exemplo, pode estar desesperado para saber se o risco de recorrência é recessivo de 25%, ou se é muito baixo devido a uma variante genética de novo.
Anormalidades identificadas na gestação
Como já mencionado, as indicações para testes invasivos incluem risco combinado aumentado no primeiro trimestre, risco aumentado após NIPT , risco aumentado na triagem bioquímica do segundo trimestre e resultados anormais de exames (por exemplo, anormalidades estruturais ou TN ≥3,5 mm no ultrasom). Estas são uma indicação para QF-PCR fetal e análise de array-CGH. Na vigência da não identificação da doença, e a depender da gravidade das estruturas comprometidas uma análise completa do exoma ou painéis específicos poderá ser necessário.
Outros fatores de alto risco
Esses fatores incluem consanguinidade parental, histórico obstétrico ruim e certas doenças maternas. A consanguinidade parental aumenta o risco de uma criança ter uma doença hereditária ou anomalia congénita . Consequentemente, se os pais estiverem preocupados, é apropriado oferecer USS detalhados para tentar excluir uma anomalia estrutural grave. Também pode ser apropriado considerar o teste de portador para quaisquer condições recessivas comuns relevantes para a etnia e história familiar do casal. Uma história obstétrica deficiente, como aborto espontâneo recorrente ou nado-morto anterior inexplicável, também é uma indicação para monitorização de futuras gestações, incluindo USS detalhada.
Uma história de três ou mais abortos inexplicáveis deve levar à análise genética para procurar um rearranjo cromossômico, como uma translocação ou inversão. É recomendado que esta análise seja realizada em produtos da concepção, com acompanhamento dos pais se os resultados sugerirem que um dos pais pode ter um rearranjo cromossômico equilibrado. Tal como acontece com os testes pré-natais, estes testes normalmente compreendem um QF-PCR seguido de array-CGH. Doenças maternas, como diabetes mellitus mal controlada ou epilepsia tratada com medicamentos anticonvulsivantes, como valproato de sódio, também são indicações para USS detalhada devido ao risco aumentado de anormalidades fetais estruturais.
Problemas especiais no diagnóstico pré-natal
O significado do resultado de um teste pré-natal é muitas vezes claro, mas podem surgir situações que colocam grandes problemas de interpretação. Problemas também ocorrem quando a investigação diagnóstica não é bem-sucedida ou quando um resultado inesperado é obtido.
Falha na obtenção de uma amostra ou falha na cultura
É importante que todas as mulheres submetidas a um destes procedimentos invasivos sejam alertadas para a possibilidade de, ser impossível obter uma amostra adequada ou de as células obtidas posteriormente não crescerem. Felizmente, o risco de ocorrência de qualquer um destes eventos é inferior a 1%.
Um resultado cromossômico ambíguo
Em aproximadamente 1% dos casos, o CVS mostra evidências de aparente mosaicismo cromossômico – isto é, a presença de duas ou mais linhagens celulares com diferentes constituições cromossômicas . Isso pode ocorrer por vários motivos:
1. A amostra está contaminada por células maternas. É mais provável que isto seja observado em células cultivadas do que em preparações diretas.
2. O mosaicismo é um artefato cultural . Geralmente, mais de uma cultura celular é estabelecida no momento do procedimento para ajudar a resolver esse problema rapidamente. Se o mosaicismo estiver presente em apenas uma cultura, então provavelmente é um artefato, não refletindo o verdadeiro cariótipo fetal.
3. O mosaicismo limita-se a uma porção da placenta, ou o que é conhecido como mosaicismo placentário confinado (CPM). Isto surge devido a um erro na mitose durante a formação e desenvolvimento do trofoblasto. Embora isto não tenha consequências para o estado cromossómico do feto, pode ter impacto na gestação porque a placenta pode não funcionar tão eficazmente, levando à restrição do crescimento no segundo e terceiro trimestres.
4. Existe um verdadeiro mosaicismo fetal .
No caso da amniocentese, na maioria dos laboratórios é rotina a realização de mais de uma cultura separada. Se uma única célula anormal for identificada em apenas uma cultura, isso é considerado um artefato de cultura, ou o que é denominado mosaicismo de nível 1 , ou pseudomosaicismo . Se o mosaicismo se estender a duas ou mais células em duas ou mais culturas, isso é considerado evidência do verdadeiro mosaicismo, ou o que é conhecido como mosaicismo de nível 3 . A situação mais difícil de interpretar é quando o mosaicismo está presente em duas ou mais células em apenas uma cultura, denominado mosaicismo de nível 2 . É mais provável que isto represente um artefato de cultura, mas há até 20% de chance de mosaicismo fetal verdadeiro.
Para resolver a incerteza do mosaicismo cromossômico em cultura de tecido CV, pode ser necessário proceder à amniocentese. Se o último teste produzir um resultado cromossômico normal, geralmente conclui-se que o resultado anterior representou CPM.
O aconselhamento nesta situação pode ser extremamente difícil. Se o verdadeiro mosaicismo for confirmado, é extremamente difícil prever o resultado fenotípico do bebê, que dependeria do nível de mosaicismo nos diferentes tecidos. Em teoria, a amostragem de sangue fetal poderia ser realizada para posterior análise cromossômica, embora isso fornecesse informações adicionais limitadas e raramente fosse realizada, dados os riscos associados. Qualquer que seja a opção escolhida pelos pais, é importante que o tecido (sangue, pele ou placenta) seja obtido no momento do parto, quer o casal decida interromper ou continuar a gestação, para resolver a importância dos resultados pré-natais.
Um resultado cromossômico inesperado
Podem ocorrer quatro tipos diferentes de resultados cromossômicos inesperados, cada um dos quais geralmente necessita de aconselhamento genético especializado e detalhado.
Uma anormalidade cromossômica numérica diferente
Embora a maioria dos procedimentos invasivos (ou seja, BVC e amniocentese) sejam realizados devido a um risco aumentado de trissomia (13, 18 ou 21) identificado como resultado da triagem no primeiro trimestre, uma anomalia cromossômica diferente dessas três trissomias pode ser encontrada , por exemplo, uma aneuploidia do cromossomo sexual (45,X, 47,XXX, 47,XXY ou 47,XYY). As aneuploidias dos cromossomos sexuais apresentam desafios de aconselhamento. É muito difícil cobrir todos os resultados possíveis do teste no momento do procedimento – mesmo os mais comuns – por isso, quando um resultado como a síndrome de Turner (45,X;) ou a síndrome de Klinefelter (47 ,XXY;), é essencial que os pais recebam detalhes completos sobre a natureza e as consequências do diagnóstico. Quando está disponível aconselhamento objectivo e informado, menos de 50% dos pais de um feto com diagnóstico “incidental” de anomalia cromossómica sexual optam pela interrupção da gestação.
Um rearranjo cromossômico estrutural
Uma segunda situação difícil é a descoberta de um rearranjo cromossômico aparentemente equilibrado no feto, como uma inversão ou translocação. Se a análise dos cromossomos parentais mostrar que um dos pais tem o mesmo rearranjo cromossômico estrutural, eles podem ter certeza de que é muito improvável que isso cause problemas na criança. Se, no entanto, este for um evento de novo no feto, há uma chance de 5% a 10% de que o feto tenha um rearranjo sutil e desequilibrado, com anormalidades físicas resultantes e/ou atraso no desenvolvimento. Este problema deve ser amplamente eliminado pelo uso de CGH-array no lugar do cariótipo no ambiente pré-natal. Array-CGH não detectará um rearranjo equilibrado, mas identificará os produtos de um rearranjo desequilibrado, desencadeando mais testes parentais. A família extensa deve ser investigada caso seja constatado rearranjo em um dos pais.
A presença de um cromossomo marcador
Outra situação difícil é a descoberta de um pequeno cromossomo “marcador” adicional, ou seja, um pequeno fragmento cromossômico cuja identidade específica não pode ser determinada por técnicas citogenéticas convencionais . Se isso estiver presente em um dos pais, é improvável que tenha algum significado para o feto, mas se for de novo , há até 15% de chance de o feto ser fenotipicamente anormal. O risco é menor quando o cromossomo marcador contém material satélite ou é composto principalmente de heterocromatina do que quando não possui satélites e é composto principalmente de eucromatina.
A disponibilidade de hibridização fluorescente in situ e array-CGH (Figura 5.7 ) significa que a origem do cromossomo marcador pode muitas vezes ser determinada de forma mais específica, o que pode ajudar na interpretação prognóstica. A anormalidade mais comum desse tipo é um marcador do cromossomo 15.
Uma descoberta incidental
Existem diretrizes uso de CGH-array pré-natal, e certos achados, sendo os loci de neurosuscetibilidade (por exemplo, deleções 15q11) um bom exemplo, não seriam relatados rotineiramente no ambiente pré-natal. No entanto, há ocasiões em que resultados considerados relevantes para o feto ou para a família seriam relatados, mesmo que não sejam a explicação para a anomalia que desencadeou o teste invasivo.
Um bom exemplo é a identificação de uma deleção do cromossomo X envolvendo o gene da distrofina em um feto do sexo feminino, confirmando assim o status de portador da distrofia muscular de Duchenne (DMD). Relatar isto permite testar a mãe, o que pode ser relevante para futuras gestações masculinas, permite o rastreio cardíaco em mulheres portadoras e é relevante quando a criança tem seus próprios filhos.
Marcadores ultrassonográficos “soft” ou sutis.
A USS sofisticada resultou na identificação de anomalias sutis no feto, cujo significado nem sempre é claro. Por exemplo, cistos do plexo coroide são algumas vezes observados nos ventrículos cerebrais em desenvolvimento no meio do trimestre (Fig. 15 ). Inicialmente, pensava-se que estes estavam invariavelmente associados ao facto de o feto ter trissomia do 18, mas na verdade ocorrem frequentemente em fetos normais, embora se forem grandes e não se resolverem espontaneamente, podem estar associados a uma anomalia cromossómica.
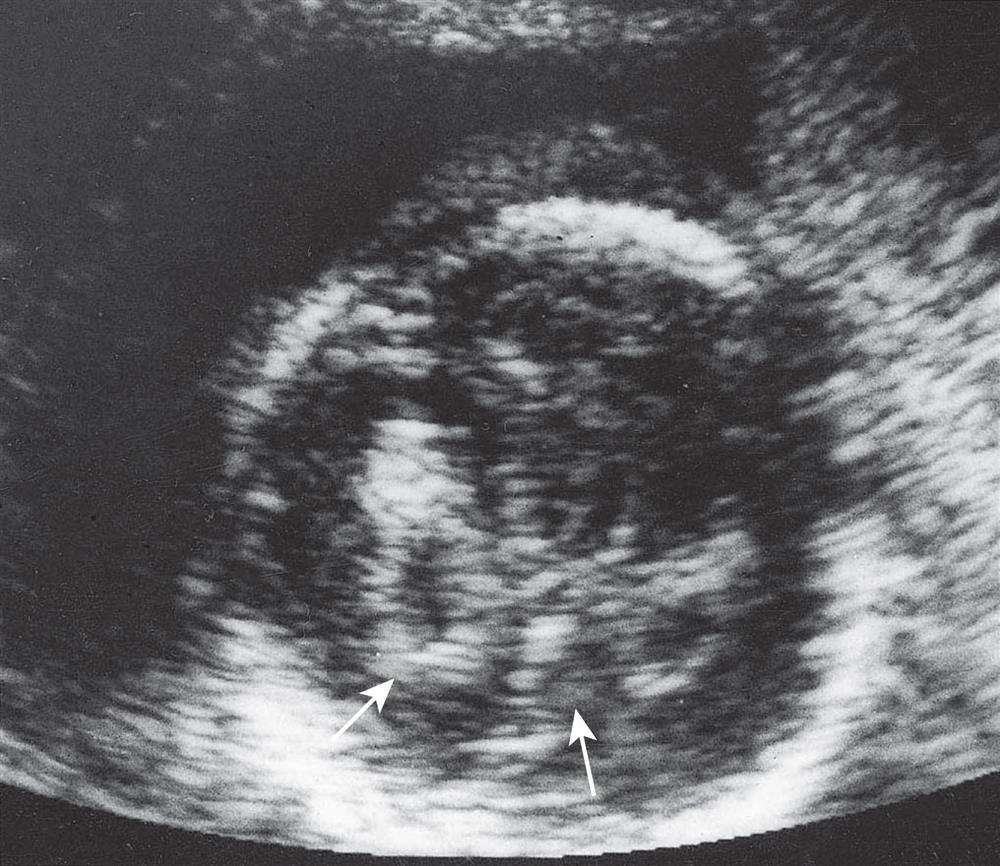
Figura 15 Ultrassonografia de um cérebro fetal
mostrando cistos bilaterais do plexo coróide (setas).
O aumento da ecogenicidade do intestino fetal (Fig. 16 ) foi relatado em associação com fibrose cística (FC) – o equivalente pré-natal do íleo meconial . Os relatórios iniciais sugeriram que esta descoberta poderia representar um risco tão elevado como 10% para o feto ter FC, mas é agora claro que este risco provavelmente não é superior a 1% a 2%. Novos achados ultrassonográficos desse tipo são frequentemente chamados de marcadores suaves , e uma abordagem cautelosa na interpretação é apropriada, incluindo exames seriados.
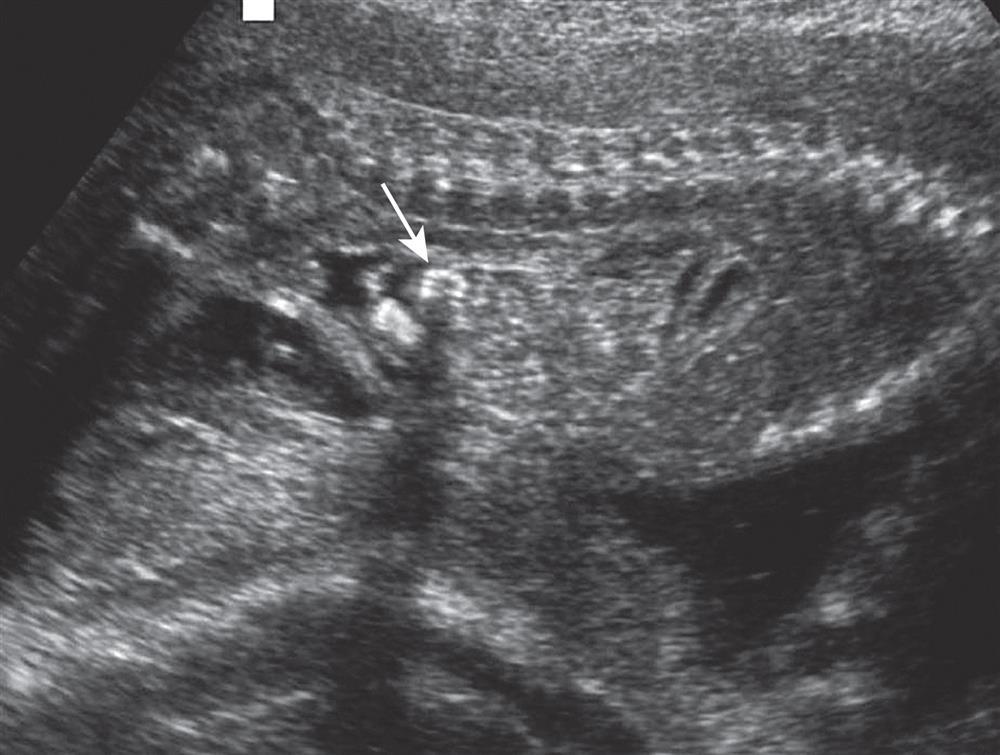
Figura 16 Intestino ecogênico. Regiões do intestino
mostrando sinal anormalmente alto (seta). Ocasionalmente, isso
é um sinal de íleo meconial observado na fibrose cística.
Interrupção da gestação
A presença de uma anomalia grave num feto em alguns países é uma normatizada para o procedimento de interrupção da gestação. No entanto, o feticídio não é consenso, e existem aspectos éticos, morais e religiosos que dispensam apresentação, e não serão abordados nesta apresentação.
Diagnóstico Genético Pré-implantacional
Para muitos casais, o diagnóstico genético pré-implantacional (DGP ou PGD) constitui uma alternativa permitida até o momento, da mesma forma é importante salientar que existem aspectos éticos, morais e religiosos que dispensam apresentação na manipulação de embriões.
No entanto para fins de informação, o segundo maior grupo de utilizadores de DGP são aqueles com baixa fertilidade (subfertilidade) ou infertilidade que desejam combinar a reprodução assistida com testes genéticos do embrião inicial.
No procedimento, a parceira recebe hormônios para induzir a hiperovulação, e os oócitos são então colhidos transcervicalmente, sob sedação e orientação ultrassonográfica.
Os espermatozoides móveis de uma amostra de sêmen são adicionados aos oócitos em cultura (em vitro fertilização [FIV] - a mesma técnica desenvolvida para infertilidade) e incubada para permitir que a fertilização ocorra - ou, comumente, a fertilização é obtida usando injeção intracitoplasmática de espermatozóide (ICSI).
No estágio de oito células (blastocisto), o embrião inicial é biopsiado e uma, ou às vezes duas, células (blastômeros) são removidas para análise. Qualquer que seja a análise genética realizada, é essencial que esta seja uma possibilidade prática no material genómico de uma única célula e, em muitos casos, uma análise utilizando um método de amplificação do genoma denominado amplificação por deslocamento múltiplo e marcadores de haplótipos - haplotipagem genética pré-implantação - que foi pioneira em 2006 é o método de escolha.
A técnica revela a origem parental dos alelos herdados e reduz a vulnerabilidade à contaminação por DNA estranho, bem como o problema de abandono de alelos, melhorando assim significativamente a eficiência. Dos embriões testados, um ou dois que sejam saudáveis e não afetados pela doença da qual correm risco são reintroduzidos no útero da mãe.
A implantação deve então ocorrer para uma gestação bem-sucedida, e este é um grande obstáculo – a taxa de sucesso do procedimento é de apenas cerca de 30% por ciclo de tratamento, mesmo nos melhores centros, embora continue a melhorar. Uma variação da técnica é a remoção do primeiro, e muitas vezes do segundo, corpo polar do ovócito não fertilizado, que fica sob a zona pelúcida. Como o primeiro corpo polar se degenera rapidamente, a análise é necessária dentro de 6 horas após a recuperação.
A análise dos corpúsculos polares é um método indireto de genotipagem porque o ovócito e o primeiro corpúsculo polar se dividem durante a meiose I e, portanto, contêm membros diferentes de cada par de cromossomos homólogos.
No os centros devem ser licenciados para praticar o PGD e são regulamentados pela Autoridade Sanitária ou de Saúde dos países, e preferencialmente Acreditados com cumprimento de normas internacionais e padrão de qualidade. Em termos numéricos, o impacto do PGD tem sido pequeno até à data. Testes para mais de 600 condições genéticas já são possivelmente realizados (exemplos na Tabela 4).
Os motivos de encaminhamento mais comuns para doenças monogênicas são FC, distrofia miotônica, doença de Huntington, β-talassemia, atrofia muscular espinhal e síndrome do X-frágil. A técnica para identificar alelos normais e anormais nestas condições e para realizar análise de ligação de DNA, quando apropriado, é a PCR .
A selecção do sexo no caso de doenças graves ligadas ao X é permitida em alguns locais quando a análise de um único gene não é possível.
O maior grupo de encaminhamentos para PGD, no entanto, são as anomalias cromossômicas – translocações recíprocas e robertsonianas em particular.
Tabela 4. Algumas condições cujo diagnóstico genético pré-implantacional está disponível
| Modo de herança | Doença |
| Autossômico dominante | Charcot-Marie-Tooth Polipose adenomatosa familiar doença de Huntington síndrome de Marfan Distrofia miotônica Neurofibromatose Osteogênese imperfeita Esclerose tuberosa BRCA1 + BRCA2 |
| Autossômica recessiva | β-Talassemia Fibrose cística Epidermólise bolhosa Doença de Gaucher Anemia falciforme Atrofia muscular espinhal Doença de Tay-Sachs |
| Ligado ao X | Síndrome de Alport Distrofia muscular de Duchenne (DMD) Síndrome de caçador Síndrome de Kennedy Síndrome do X Frágil |
| Ligado ao X: apenas sexagem | DMD Deficiência de ornitina transcarbamilase Incontinência pigmentar |
| Mitocondrial | MELAS |
| Cromossômico | Translocações Robertsonianas Translocações recíprocas Inversões, exclusões |
MELAS, Miopatia mitocondrial, encefalopatia, acidose láctica, acidente vascular cerebral.
Nos últimos anos, o PGD tem sido utilizado, em raras ocasiões, não só para seleccionar embriões não afectados pela doença genética para a qual a gestação está em risco, mas também para fornecer uma correspondência do tipo de tecido do antigénio leucocitário humano, para que a nova criança possa actuar como um doador de medula óssea para um irmão mais velho afetado, por exemplo, por anemia de Fanconi.
Um desenvolvimento adicional usando métodos de micromanipulação atraiu muita atenção. Para contornar o problema da doença genética devastadora resultante de uma variante patogénica no genoma mitocondrial (onde o risco de recorrência pode chegar a 100%), o núcleo do ovócito da mãe genética (portadora da variante mitocondrial) pode ser removido e inserido em um ovócito doador do qual o núcleo foi removido. Esta é uma tecnologia de substituição nuclear celular, semelhante à utilizada em experiências de clonagem reprodutiva em animais (“Dolly” a ovelha; ver). A controvérsia ética foi alimentada pela frase de efeito mediática “bebês com três pais”, embora o DNA do doador represente 0,005% do total. Parte da preocupação está relacionada com o potencial de transmissão matrilinear das mitocôndrias doadoras para as gerações futuras.
Concepção Assistida e Implicações para Doenças Genéticas
Fertilização in vitro
Muitos bebês em todo o mundo nasceram por fertilização in vitro desde 1978, quando a técnica levou ao primeiro nascimento vivo. A indicação do tratamento na maioria dos casos é a subfertilidade, que hoje atinge um em cada sete casais. Em alguns países ocidentais, 1% a 3% de todos os nascimentos resultam de tecnologias de reprodução assistida (TARV).
A estimulação controlada dos ovários com gonadotrofinas exógenas leva ao recrutamento de muitos folículos (monitorizados por USS). Quando o número e o tamanho dos folículos em desenvolvimento são considerados apropriados, a maturação dos oócitos é desencadeada hormonalmente. Trinta e quatro a 38 horas depois, os oócitos são recolhidos por aspiração guiada por USS transvaginal do fluido folicular. Os oócitos são então fertilizados in vitro com o espermatozoide do parceiro e quaisquer embriões morfologicamente sãos resultantes são transferidos para o útero da mulher 2–7 dias depois.
A coorte de descendentes concebidos desta forma é, portanto, grande, e estão a reunir-se evidências de que o risco de defeitos congénitos aumenta entre 30% a 40% em comparação com a população geral concebida naturalmente, com cerca de 50% mais crianças susceptíveis de serem pequenas para a idade (PIG) com OR de até 1,7 ou seja, até 70% a mais que a população em geral. Cerca de 7 à 8% dos bebés concebidos por FIV podem apresentar um defeito congénito diagnosticado antes do quinto ano de vida, comparado com cerca de 6% nos bebés concebidos sem tratamento de concepção assistida.
Especificamente, foi observado um pequeno aumento em certas condições epigenéticas, cerca de um em 4000, devido à impressão genômica defeituosa maternal associado as síndromes de Beckwith-Wiedemann e Angelman e mais raramente as síndrome de “hipometilação”, incluindo as síndromes como Prader-Willi e Silver-Silver associadas com alterações no imprinting paternal respectivamente no mesmo locus de Angelman e no gene H19, no cromossomo 11 . Nos casos estudados, foi observada perda de imprinting no locus KCNQ1OT1 no caso da síndrome de Beckwith-Wiedemann e no locus SNRPN no caso de Síndrome de Angelman. Os mecanismos para estes fenômeno ainda não estão bem estabelecidos.
Os eventos epigenéticos na época da fertilização e implantação são cruciais para o desenvolvimento normal . Se houver um risco claramente aumentado de condições resultantes de impressões anormais após as TARV, isto pode estar relacionado, em parte, com o tempo prolongado de cultura dos embriões, que se tornou uma tendência nas clínicas de infertilidade. Em vez de transferir embriões em fase de clivagem, é agora mais rotineiro transferir blastocistos, o que permite a selecção de embriões com aspecto mais saudável. No entanto, em modelos animais foi demonstrado que a cultura in vitro afecta a extensão da impressão e da expressão genética e, portanto, o potencial para o desenvolvimento normal.
Injeção intracitoplasmática de esperma (ICSI)
A ICSI envolve a injeção de um único espermatozoide diretamente no citoplasma de um oócito maduro. Esta técnica é comumente usada como parte da fertilização in vitro quando combinada com PGD, mas a principal indicação para a injeção direta de espermatozóides no óvulo é a subfertilidade masculina devido à baixa contagem de espermatozoides, baixa motilidade espermática, morfologia anormal do esperma ou bloqueio mecânico do esperma à passagem dos espermatozoides ao longo dos canais deferentes. Além disso, a ICSI é usada para técnicas de PGD baseadas em PCR para evitar o risco de extrair espermatozoides extra, enterrados na zona pelúcida, na biópsia de embriões, o que contaminaria o ensaio.
Foram encontrados anormalidades ou rearranjos cromossômicos em cerca de 5% dos homens para os quais a ICSI é adequada e em 10% a 12% daqueles com azoospermia ou oligospermia grave. Os exemplos incluem a translocação Robertsoniana 13:14 e as deleções do cromossomo Y. Para homens com azoospermia ou oligospermia grave, o cariótipo deve ser verificado, incluindo a aplicação de técnicas moleculares em busca de deleções submicroscópicas do Y.
Naqueles com bloqueio mecânico devido à ausência bilateral congênita dos canais deferentes (CBAVD), uma proporção significativa tem variantes de FC. A ICSI oferece esperança aos homens com CBAVD, bem como àqueles com síndrome de Klinefelter, após aspiração testicular de esperma.
Algumas das anomalias cromossómicas nos homens podem ser hereditárias – especialmente aquelas que envolvem os cromossomas sexuais – e há um pequeno mas definitivo aumento nas anomalias cromossómicas na descendência (1,6%). Cerca de 10% dos bebés concebidos com ICSI tinham um defeito congénito antes do quinto aniversário da criança, comparado com até 6% nos bebés concebidos sem tratamento de conceção assistida.
Inseminação de Doadores
Como meio de concepção assistida para tratar a infertilidade masculina ou para contornar o risco de uma doença genética, a inseminação de doadores (DI) tem sido utilizada desde a década de 1950. Contudo, apenas recentemente é que a consciência das questões genéticas médicas foi incorporada na prática. Após os casos de crianças concebidas por ID que foram posteriormente descobertas como tendo distúrbios cromossômicos equilibrados ou desequilibrados, ou em alguns casos FC (indicando que o doador de esperma era portador de FC), a triagem de doadores de esperma para variantes de FC e rearranjos cromossômicos tornou-se prática rotineira em muitos países. Isto foi recomendado apenas recentemente, em 2000, pela Sociedade Britânica de Andrologia. Na Holanda, um doador cujo esperma foi utilizado para gerar 18 descendentes desenvolveu uma doença neurodegenerativa de início tardio da DA (uma das ataxias espinocerebelares), indicando assim que todos os 18 descendentes concebidos corriam 50% de risco. Isto levou a uma decisão de que o esperma de um doador não deveria ser usado mais de 10 vezes, contra 25 antes desta experiência. Homens com mais de 40 anos apresentam risco, pequeno mas crescente, de novas variantes germinativas surgirem no esperma com o avanço da idade paterna.
É claro que não é possível rastrear o dador para todas as eventualidades, mas estes casos serviram para realçar o conflito potencial entre o tratamento da infertilidade (ou doença genética) por DI e a manutenção de um elevado nível de preocupação com o bem-estar da criança concebida. Mais importante a este respeito é o debate em curso sobre quanta informação as crianças DI devem ter sobre os seus pais genéticos, e a lei varia em todo o mundo. As questões aplicam-se igualmente às mulheres que doam os seus óvulos.
Aspectos Legais
Nos Estados Unidos, não existe nenhuma lei federal para regular a prática da concepção assistida, exceto a exigência de que os resultados da fertilização in vitro e da ICSI sejam relatados. No Reino Unido, existe uma regulamentação rigorosa através da HFEA com base na Lei de Fertilização Humana e Embriologia de 1990 (atualizada em 2008). As diferentes licenças concedidas são para tratamento (Quadro 2 ), armazenamento (gametas e embriões) e investigação (em embriões humanos in vitro). Deve ser mantido um registro de todos os ciclos de tratamento, das crianças nascidas por fertilização in vitro e da utilização de gametas doados. A pesquisa permitida sob licença abrange o tratamento da infertilidade, o aumento do conhecimento sobre defeitos congênitos, aborto espontâneo, testes genéticos em embriões, o desenvolvimento do embrião inicial e o tratamento potencial de doenças graves.
Tratamentos de concepção assistida que exigem licença da Autoridade de Fertilização Humana na maiorias dos locais
Fertilização in vitro
Injeção intracitoplasmática de esperma
Diagnóstico genético pré-implantacional
Tratamento de doação mitocondrial
Doação de esperma
Doação de óvulos
Doação de embriões
Barriga de aluguel
Teste pré-natal não invasivo
Na virada do século XIX, descobriu-se que as células fetais alcançavam a circulação materna, mas a confirmação de que o DNA fetal livre de células (cffDNA), derivado do tecido trofoblástico da placenta, está presente no plasma de mulheres grávidas só ocorreu após o final da década de 90 (Fig. 17 ). Evidencias demonstraram que na 6 ª a 7ª semanas de gestação é possível determinar o sexo fetal através da detecção de sequências de DNA do cromossoma Y e do gene Rhesus D fetal. A determinação precoce do sexo fetal é clinicamente útil numa gestação com risco de doença recessiva ligada ao X e permite uma redução de 50% na necessidade de testes invasivos. O problema com a análise do cffDNA é o isolamento porque o DNA livre de células maternas constitui 80% a 90% de todo o DNA livre de células na circulação materna. A ausência de DNA do cromossomo Y pode indicar que o feto é do sexo feminino ou que a quantidade de DNA fetal é muito baixa. Isto é resolvido usando PCR em tempo real para quantificar a quantidade de DNA fetal ou total presente no plasma.
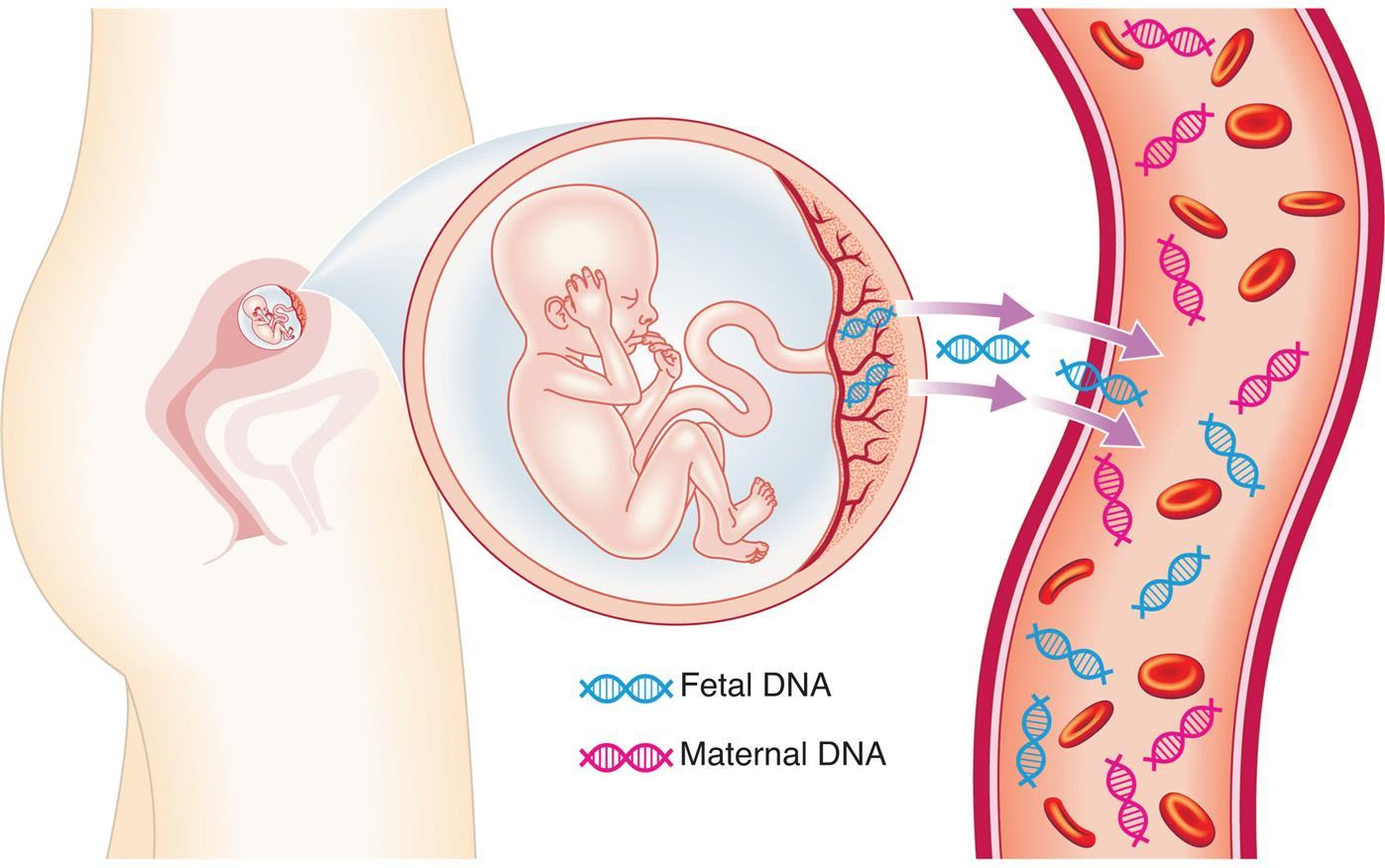
Figura 17 Pequenas quantidades de DNA fetal livre de
células chegam à circulação materna a partir dos trofoblastos da
placenta e podem ser acessadas para análise genética.
O desenvolvimento da tecnologia para detectar a síndrome de Down e outras trissomias comuns no feto tem sido rápido. Neste caso, o desafio reside em discriminar entre uma amostra de DNA em que o componente fetal constitui três cópias do cromossoma 21, em oposição às duas cópias do DNA livre de células plasmáticas maternas. Isto foi conseguido usando tecnologia massiva de sequenciamento paralelo “shotgun” combinada com sofisticada análise de dados de sequenciamento. Essencialmente, milhões de pequenos fragmentos de cffDNA (tanto aleatórios quanto específicos dos cromossomos de interesse) do plasma materno (contendo DNA livre de células fetais e maternas) são amplificados e sequenciados. Os fragmentos são então mapeados no genoma humano e analisados quanto à sua frequência, ou densidade, ao longo de cada cromossomo, permitindo a detecção da síndrome de Down no feto, onde os fragmentos do cromossomo 21 estão super-representados, e da mesma forma para outras aneuploidias comuns. Estudos demonstraram uma precisão de 99% para a trissomia 21, 96% para a trissomia 18 e 91% para a trissomia 13.
Conforme discutido anteriormente , substituir o actual rastreio precoce da gestação apenas pelo NIPT seria dispendioso e à custa do benefícios das metodologias de triagem atuais. No entanto, o NIPT tornar-se-á, sem dúvida, uma parte essencial do rastreio de rotina, em combinação com os testes já disponíveis, e foi calculado como tendo uma boa relação custo-eficácia quando utilizado para substituir CVS/amniocentese após um resultado de teste combinado de alto risco no primeiro trimestre (≥ 1:150 risco de trissomia). É importante lembrar que o NIPT continua a ser um teste de rastreio e não deve ser utilizado para confirmar um diagnóstico de trissomia. Um resultado NIPT de alto risco ainda requer confirmação com testes invasivos.
Algumas empresas que oferecem o NIPT também incluem avaliação de CNVs cromossômicas comuns, por exemplo, deleções 22q11. A evidência e a precisão do NIPT para este fim são menos claras e, tais testes só estão disponíveis através de estudos de investigação ou quando autofinanciados de forma particular.
Diagnóstico pré-natal não invasivo
Foi um grande avanço clínico a possibilidade de um exame pré-natal muito preciso, que evite um procedimento invasivo com risco de perda fetal. Como resultado, a avaliação não invasiva do cffDNA expandiu-se, permitindo o teste de uma série de condições genéticas. Isto está disponível para Fibrose Cística , síndrome de Apert (FGFR2 -), displasia esquelética relacionada ao FGFR3 , DMD e distrofia muscular de Becker , displasia muscular espinhal atrofia , hiperplasia adrenal congênita e algumas das condições de craniossinostose secundárias a variantes do FGFR2, entre outras.
Além disso, o diagnóstico pré-natal não invasivo personalizado também é possível com testes concebidos individualmente para a doença e variantes genéticas específicas que afetam uma família. Para condições recessivas, isto geralmente envolverá a procura de evidências da variante do gene patogénico paterno, que, se ausente, seria tranquilizadora, uma vez que o bebé poderia ser apenas um portador da doença. Quando a variante paterna for identificada, serão oferecidos aos casais testes invasivos para confirmar se o feto é apenas portador ou afetado pela doença. O uso do NIPD (ou NIPT) reduzirá a necessidade de testes pré-natais invasivos, de forma semelhante à sexagem fetal para doenças ligadas ao cromossomo X.
Embora existam preocupações inevitáveis de que a tecnologia tornará possível testar o feto relativamente a características ou características não médicas, isto é extremamente improvável, dada a natureza personalizada de cada ensaio individual. No entanto, muda drasticamente a face dos testes e rastreios pré-natais num futuro próximo.
Tabela 5. Alguns distúrbios genéticos para os quais o PGD está atualmente disponível:
| Autossômico dominante: |
|---|
| • Distrofia miotônica |
| • Doença de Huntington (para portadores de mutação conhecida e também teste de exclusão para aqueles com 50% de risco de ter uma mutação) |
| • BRCA1 e 2 |
| Autossômico recessivo |
| • Fibrose cística |
| • Atrofia muscular espinhal |
| • Beta-talassemia |
| • Doença falciforme |
| Ligado ao cromossomo X |
| • Síndrome do X frágil |
| • Síndrome de Alport |
| • Distrofia muscular de Duchenne/Becker |
| • Hemofilia |
| • Síndrome de Hunter |
| • Incontinentia pigmenti |
| Distúrbios cromossômicos‡ |
| • Translocações robertsonianas |
| • Translocações recíprocas |
| • Outros distúrbios cromossômicos (inversões, deleções) |
‡ Dependente da disponibilidade de sondas FISH.
Dados da Human Fertilisation and Embryology Authority (HFEA) http://guide.hfea.gov.uk/pgd/.
Sequenciamento rápido pré-natal do exoma
Uma das dificuldades da genética pré-natal é fazer o diagnóstico em um período limitado de tempo, com detalhes fenotípicos limitados. Como testes genéticos extensos, por exemplo um grande painel genético, podem levar muitos meses para serem concluídos, grande parte do aconselhamento pré-natal será baseado em resultados de exames e em possíveis diagnósticos genéticos, talvez apenas conseguindo confirmar o diagnóstico mais tarde na gestação.
Uma opção, ou após a conclusão da gestação. Para as gestações que apresentam um quadro complexo de anomalias congénitas, com testes cromossómicos normais e onde se pensa que um diagnóstico genético é provável, a sequenciação do exoma está a tornar-se uma parte fundamental do caminho diagnóstico. O teste rápido, que fornece resultados em algumas semanas, pode ser extremamente benéfico para um casal, pois fornece informações sobre o prognóstico do feto. Em alguns casos, por exemplo, certas condições metabólicas, os resultados permitiram um tratamento rápido no recém-nascido. Nos próximos anos, a sequenciação do genoma completo terá inevitavelmente um papel também no cenário pré-natal e, embora muitas questões envolvam a utilização e interpretação de dados tão complexos, os benefícios poderão ser significativos neste campo da genética.
Tratamento pré-natal
Existem estudos de transplante pré-natal de células-tronco em fetos com diagnóstico de osteogênese imperfeita, que resultou em uma redução no número de fraturas. O tratamento de um feto afetado por imunodeficiência combinada grave também foi relatado. A tolerância imunológica do feto a antigénios estranhos introduzidos no útero significa que as células estaminais transfundidas são reconhecidas como “próprias”, com a perspectiva de bons resultados a longo prazo.
Quando a terapia genética se mostrar segura e eficaz, a tolerância imunológica do feto deverá facilitar o início dessa terapia antes do nascimento, e não depois. Isto terá a vantagem adicional de reduzir o período em que podem ocorrer danos irreversíveis em órgãos como o sistema nervoso central, que pode ser afetado por doenças neurodegenerativas progressivas.
Resumo
1. A triagem pré-natal pode ser realizada por métodos não invasivos, como a triagem combinada do primeiro trimestre para as síndromes de Down, Edwards e Patau, que combina marcadores bioquímicos, medição da translucência nucal e idade materna. A ultrassonografia detalhada para anormalidades estruturais é uma parte vital da triagem pré-natal.
2. Testes pré-natais específicos de distúrbios cromossômicos e de genes únicos tradicionalmente dependiam de técnicas invasivas, como amniocentese ou amostragem de vilosidades coriônicas, para obter material de origem fetal para análise. Embora estes continuem a ser testes frequentemente solicitados, abordagens não invasivas estão a tornar-se cada vez mais disponíveis.
3. Procedimentos invasivos de testes pré-natais apresentam pequenos riscos de causar aborto espontâneo (por exemplo, amniocentese 0,5% a 1%, amostragem de vilosidades coriônicas 1%, cordocentese 1% a 2%, fetoscopia 3% a 5%).
4. As indicações comuns para testes pré-natais invasivos incluem: risco combinado aumentado ou translucência nucal elevada, história anterior ou familiar de distúrbio cromossômico ou de gene único ou anormalidades estruturais identificadas na ultrassonografia.
5. Embora o significado de muitos resultados de diagnóstico pré-natal seja claro, surgem frequentemente situações em que as implicações para o feto são muito difíceis de prever, caso em que o casal deve receber aconselhamento genético especializado.
6. Os testes pré-natais não invasivos estão a mudar a abordagem do rastreio pré-natal e proporcionam maior precisão nos testes das trissomias comuns, mas não são considerados confirmativos do diagnóstico.
7. O diagnóstico pré-natal não invasivo está cada vez mais disponível para um espectro de doenças monogênicas, reduzindo assim a necessidade de testes invasivos.
8. A sequenciação completa do exoma e do genoma completo está a começar a desempenhar um papel no diagnóstico pré-natal de presumíveis doenças genéticas e é provável que tenha um impacto significativo nos próximos anos.
9. A tecnologia não só avançou em termos de testes durante a gestação, mas o sucesso crescente do diagnóstico genético pré-implantação está a tornar esta uma escolha popular para muitos casais que desejam evitar os testes pré-natais.
Cenário Clínico 1
Uma mulher nulípara de 36 anos recebe um resultado de triagem combinada de alto risco com base na idade materna e uma baixa dosagem de proteína A plasmática associada à gestação (<0,4 múltiplos da mediana) às 12 semanas de gestação. Nenhuma outra anormalidade na varredura é detectada.
Quais são as opções de gerenciamento disponíveis? Como os resultados de mais testes impactariam o manejo da gestação?
Cenário Clínico 2
Um casal é encaminhado à clínica de genética com base na história familiar de fibrose cística. Eles estão esperando o primeiro filho, atualmente com 6 semanas de gestação.
Como você os aconselharia sobre o risco para o feto, que testes adicionais você pode oferecer e que testes estão disponíveis durante a gestação, caso ambos os pais sejam portadores?
Leitura adicional
Allyse et al., 2015 Allyse M, Minear MA, Berson E, et al. Testes pré-natais não invasivos: uma revisão da implementação e dos desafios internacionais. Int J Saúde da Mulher . 2015;7:113–126.
Uma revisão da introdução internacional do NIPT e dos desafios da implementação da sua utilização em todo o mundo .
Best et al., 2018 Best S, Wou K, Vora N, et al. "Promessas, armadilhas e aspectos práticos do sequenciamento completo do exoma pré-natal" . Pré-diagnóstico . 2018;38(1):10–19.
Uma revisão dos estudos pré-natais de sequenciamento completo do exoma: as taxas de diagnóstico, desafios e limitações .
Evans, 2006 Evans M. Diagnóstico pré-natal McGraw-Hill Professional 2006.
Uma referência abrangente sobre diagnóstico e avaliação de riscos reprodutivos e gestações de alto risco geneticamente relacionadas .
Ghi et al., 2016 Ghi T, Sotiriadis A, Calda P, et al. Diretrizes Práticas ISUOG: procedimentos invasivos para diagnóstico pré-natal. Ultrassonografia Obstet Gynecol . 2016;48(2):256–268.
Uma diretriz que resume quando, como e por que realizar procedimentos invasivos para diagnóstico pré-natal .
Gil et al., 2015 Gil MM, Quezada MS, Revello R, Akolekar R, Nicolaides KH. "Análise de DNA livre de células no sangue materno na triagem de aneuploidias fetais: meta-análise atualizada". Ultrassonografia Obstet Gynecol . 2015;45:249–266.
Uma meta-análise do desempenho do teste de cfDNA na triagem de trissomias fetais 21, 18, 13 e aneuploidias dos cromossomos sexuais .
Autoridade de Fertilização Humana e Embriologia Autoridade de Fertilização Humana e Embriologia. Condições aprovadas para PGD . Disponível em: https://www.hfea.gov.uk/pgd-conditions/ .
Uma lista de condições aprovadas e aquelas que aguardam aprovação do HFEA, para diagnóstico genético pré-implantação .
Comitê Conjunto de Genômica em Medicina, Comitê Conjunto de Genômica em Medicina de 2015. Recomendações para o uso de microarranjos cromossômicos na gestação Londres: The Royal College of Pathologists;; 2015; Disponível em: https://www.rcpath.org/uploads/assets/06664c28-0f90-4230-86158c91fea14be6/Recommendations-for-the-use-of-chromosome-microarray-in-pregnancy.pdf .
Recomendações do Reino Unido sobre as indicações e relatos de CGH-array no ambiente pré-natal .
Lord et al., 2019 Lord J, McMullan DJ, Eberhardt RY, et al. "Análise de sequenciamento de exoma pré-natal em anomalias estruturais fetais detectadas por ultrassonografia (PAGE): um estudo de coorte". Lanceta . 2019;393(10173):747–757.
Um estudo de coorte avaliando o uso de sequenciamento de exoma completo para detectar a presença de variantes em genes de distúrbios do desenvolvimento em fetos com anomalias estruturais .
Odibo e Krantz, 2010 Odibo A, Krantz D. Triagem e diagnóstico pré-natal, uma edição de clínicas em medicina laboratorial Saunders 2010.
Um texto abrangente que abrange a triagem pré-natal e o diagnóstico de doenças genéticas durante a gestação .
Pereira e Soria, 2010 Pereira E, Soria J, eds. Manual de métodos de diagnóstico pré-natal, questões e impactos na saúde . Nova Science Publishers Inc 2010.
Um texto de referência sobre diagnóstico pré-natal .
Santorum et al., 2017 Santorum M, Wright D, Syngelaki A, et al. Precisão do teste combinado do primeiro trimestre na triagem de trissomias 21, 18 e 13. Ultrassonografia Obstet Gynecol . 2017;49(6):714–720.
Um estudo prospectivo de validação de triagem para trissomias 21, 18 e 13 usando o teste combinado do primeiro trimestre .
As principais referências, consensos e diretrizes sobre reprodução humana e aconselhamento pré-natal e pré-concepcional encontram-se nas Sociedade Europeia de Reprodução Humana e Embriologia
( https://www.eshre.eu/Guidelines-and-Legal )
e na A Sociedade Americana de Medicina Reprodutiva ( https://www.asrm.org/practice-guidance ).